

Physiopathologie des maladies neuromusculaires
Chef d'équipe : Jocelyn LAPORTE
Département : Médecine translationnelle et neurogénétique
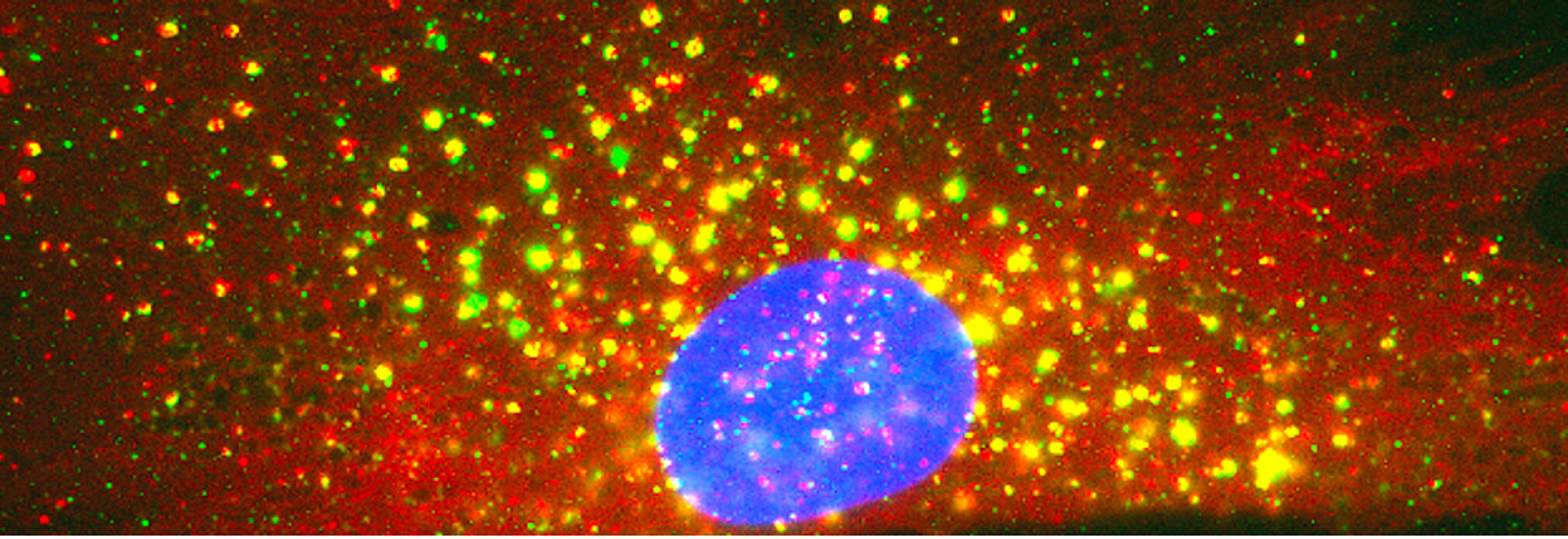
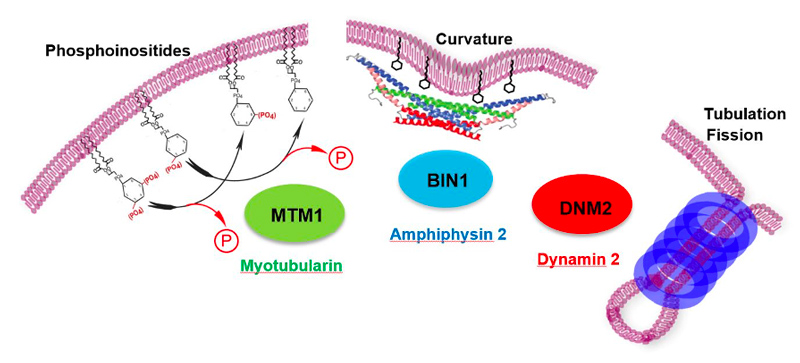
Centronuclear and myotubular myopathies (CNM) are rare and severe genetic myopathies with a strong burden on patients, their families and our healthcare system. They associate muscle weakness and hypotonia, respiratory distress and abnormal organelles positioning in myofibers. We previously identified 3 main genes mutated in CNM, all encoding proteins that regulate membrane and cytoskeleton dynamics: the phosphoinositides phosphatase myotubularin (MTM1), the membrane remodeling amphiphysin 2 (BIN1), and the membrane fissioning GTPase dynamin 2 (DNM2).
We now aim to
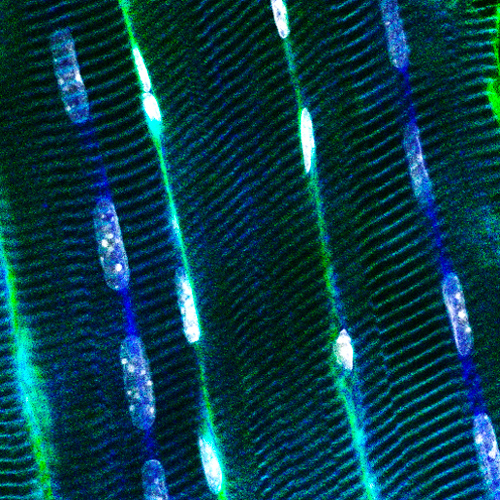
Muscle fibers can contract, are syncytia from the fusion of myoblasts, can have several hundred nuclei, measure up to 30cm in human, and account for nearly half of the dried body weight. They have specific intracellular organization and specialized membrane structures. I and my team identified the Myotubularin family (Laporte et al. 1998) and showed these proteins act mainly as phosphoinositides phosphatases regulating autophagy (Blondeau et al. 2002; Vergne et al. 2009), misfolded proteins degradation (Gavriilidis et al. 2018), reticulum remodelling (Amoasii et al. 2013) and phosphoinositides conversion in recycling (Ketel et al. 2016). Several members are mutated in neuromuscular diseases (MTM1, MTMR2, MTMR5, MTMR13). We also revealed how different proteins mutated in centronuclear myopathies (myotubularin-MTM1, and amphiphysin-BIN1) control focal adhesion (Lionello et al. 2019) and nuclear (D’Alessandro et al. 2015) or mitochondria (Hnia et al. 2011) positioning through the interaction with nesprin and desmin, two other proteins mutated in myopathies, linking several myopathies into a common cellular pathway.
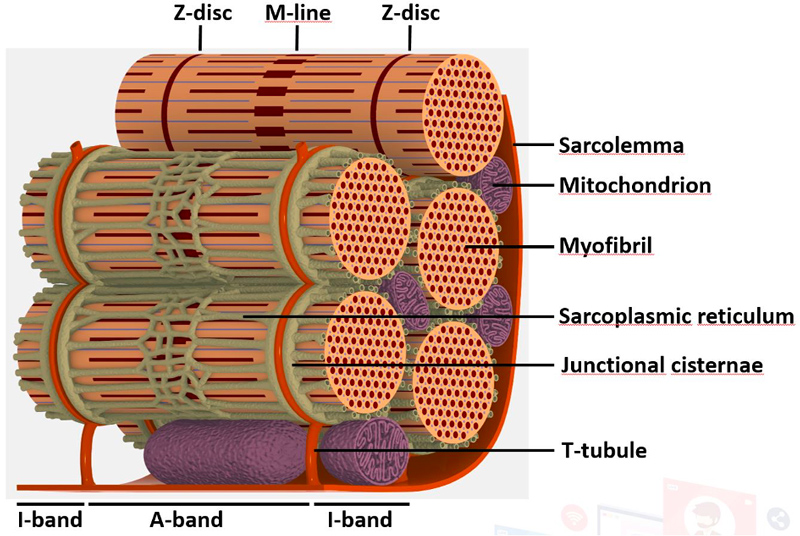
To decipher the pathological mechanisms leading to several congenital myopathies, we have characterized 14 novel murine models and identified 3 spontaneous canine models (Beggs et al. 2010; Bohm et al. 2013). We found centronuclear myopathies are linked to defects of organelle positioning and of the triads, the membrane compartment regulating excitation-contraction coupling (AlQusairi et al. 2009; Toussaint et al. 2011). In tubular aggregate myopathies, we discovered that mutations in several components of the store-operated calcium entry (SOCE) pathway regulating calcium homeostasis lead to constitutive activated SOCE and higher calcium entry (Bohm et al. 2013).
Most myopathies have no therapy. We identified 5 therapeutic targets and 1 potent drug. Using faithful cellular and animal models and the generated knowledge on the pathomechanisms, we deciphered which functions of myotubularin-MTM1 is necessary for rescuing a mouse model for myotubular myopathy through gene therapy (Amoasii et al. 2012) and showed expression of a close MTM1 homolog, MTMR2, can also rescue the disease (Raess et al. 2017). We validated the proof-of-concept that upregulation of BIN1 encoding amphiphysin 2 can prevent the development of myotubular myopathy due to MTM1 mutations (Lionello et al., 2019). Similarly, inhibition of the kinase activity of PI3K C2beta can also rescue this disease in mice (submitted). We also found tamoxifen, a drug used for breast cancer treatment, ameliorates myotubular myopathy (Gayi et al. 2018). We showed that downregulation of dynamin-DNM2 can prevent and revert myotubular myopathy in mammals through genetic cross, oligonucleotide-mediated or short hairpin RNA interference (Cowling et al. 2014; Tasfaout et al. 2017, 2018) and can be applied to several forms of centronuclear myopathies (Cowling et al. 2017; Buono et al. 2018). This last finding allowed the creation of the biotech company Dynacure end 2016 (www.dynacure.com), aiming to treat different rare diseases.

Chef d'équipe : Jocelyn LAPORTE
Département : Médecine translationnelle et neurogénétique

A l'occasion de la 36è édition du Téléthon, les scientifiques Jocelyn Laporte, Johann Böhm et Delphine Duteil ont présenté leurs projets de recherches…
Lire la suite
Proceedings of the National Academy of Sciences ; Volume: 119
JCI Insight ; Volume: 7
Nature Communications ; Volume: 13 ; Page: 2306
Proceedings of the National Academy of Sciences of the United States of America ; Volume: 119
Disease Models & Mechanisms ; Volume: 15 ; Page: dmm049219
International Journal of Molecular Sciences ; Volume: 23 ; Page: 6968
Neuromuscular Disorders ; Volume: 32 ; Page: S55
Disease Models & Mechanisms ; Volume: 15 ; Page: dmm049284
Acta Neuropathologica Communications ; Volume: 9 ; Page: 155
International Journal of Molecular Sciences ; Volume: 22 ; Page: 11377
Page 4 sur 23