
Bases Moléculaires de la Régulation de la Chromatine et de la Transcription
Bases Moléculaires de la Régulation de la Chromatine et de la Transcription

Notre équipe étudie les mécanismes épigénétiques pour comprendre leur implication dans les processus nucléaires, leurs dysfonctionnements dans les maladies, et comment moduler l'activité des effecteurs épigénétiques à l'aide de candidats médicaments.
NOS INTÉRÊTS
Comprendre les bases moléculaires des mécanismes épigénétiques
Comprendre la régulation des processus nucléaires par les effecteurs épigénétiques
Comprendre la dérégulation des mécanismes épigénétiques dans les maladies
Comprendre comment moduler l'activité des effecteurs épigénétiques par des candidats médicaments
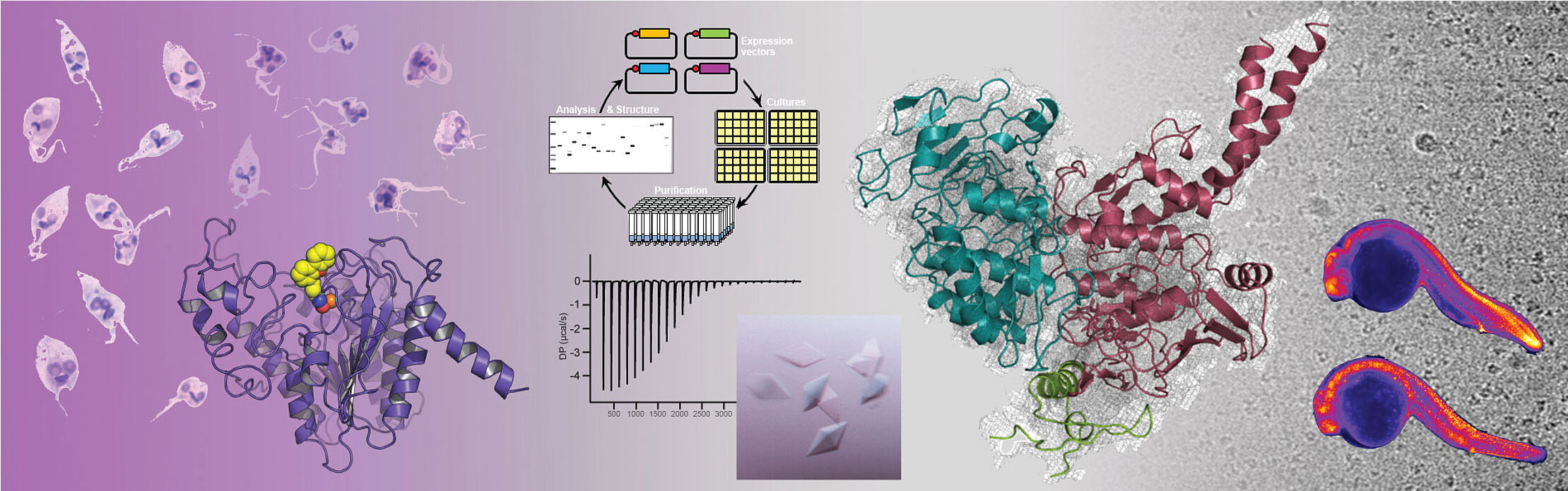
Notre équipe étudie les mécanismes épigénétiques eucaryotes qui sont essentiels à l’homéostasie cellulaire et au développement des organismes. Nous caractérisons notamment les bases moléculaires des mécanismes épigénétiques pour déchiffrer leur implication dans la régulation des processus nucléaires et comprendre leurs rôles physiologiques. Nous utilisons cette connaissance pour déterminer comment les mutations des effecteurs épigénétiques peuvent conduire à de nombreuses pathologies différentes. Nous mettons aussi en œuvre des stratégies pour caractériser de petites molécules qui inhibent ou régulent l’activité des effecteurs épigénétiques, dans le but d’aider au développement de candidats-médicaments ciblant les pathologies épigénétiques, notamment des cancers, des maladies neurodéveloppementales et des maladies négligées (malaria, leishmaniose, schistosomiase, maladie de Chagas).
NOS OBJECTIFS
Nos objectifs sont de combiner nos différents axes de recherche pour aider à la compréhension des mécanismes épigénétiques, leurs implications dans les pathologies, et participer au développement de thérapies épigénétiques. Nos études s’appuient sur des analyses in vitro biochimiques, biophysiques et de biologie structurale. Cependant, nous développons l’ensemble de nos projets dans le cadre de stratégies de biologie intégrative qui allient analyses in vitro, in cellulo et in vivo. Nous développons ainsi l’utilisation du poisson-zèbre au sein de l’équipe pour adresser nos trois axes de recherche (mécanismes, pathologies et petites molécules) dans un modèle animal pour caractériser les relations structure/fonction de nos cibles épigénétiques.
NOS PROJETS
Nos projets scientifiques ciblent particulièrement trois types d’effecteurs épigénétiques : les désacétylases, les chaperons d’histones et les complexes SMC (Structural Maintenance of Chromosomes). Nos cibles épigénétiques sont toutes reliées fonctionnellement, notre but étant à terme de caractériser leurs interrelations physiques et fonctionnelles. En soutien de nos projets scientifiques en épigénétique, notre équipe développe depuis plus de deux décades une technologie de reconstitution de complexes protéiques par multi-expression dans les hôtes bactériens. Cette technologie et notre savoir-faire associé sont essentiels à l’ensemble de nos projets scientifiques et sont aussi à la source de projets collaboratifs spécifiques avec d’autres équipes de recherche.
Membres
Chercheur(euse)s
Doctorant(e)s
Ingénieur(eure)s
Technicien(ne)s
Anciens membres
Doctorants
Pauline LANDWERLIN (2018-2022)
Marina VITORIA GOMES (2017-2021)
Elizabeth RAMOS MORALES (2017-2021)
Pernelle KLEIN (2016-2020)
Tajith Baba SHAIK (2014-2018)
Post-doctorants
Nataliia ALEKSANDROVA (2016-2018)
Martin MAREK (2010-2017)
Régis BACK (2015-2017)
Michael KOCH (2007-2009)
Sous-groupe(s)

Chimie biologique des modifications post-traductionnelles dans l’organisation de la chromatine
Responsable de sous-groupe : Christophe DECROOS
Mécanismes moléculaires de la réorganisation de la chromatine
Responsable de sous-groupe : Christophe ROMIER
Projets en cours
Désacétylases d'histones (HDAC)
L’acétylation des protéines est une modification post-translationnelle qui régule l’activité d’un très grand nombre d’acteurs cellulaires et qui rivalise en importance avec la phosphorylation. L’acétylation joue notamment un rôle essentiel en épigénétique en agissant sur la compaction de la chromatine mais aussi dans les signalisations épigénétiques. La marque d’acétylation est réversible, étant déposée par des lysines acétyl transférases (KATs), lue par des bromodomaines, et enlevée par des lysines désacétylases (KDACs). Les enzymes du cycle de l’acétylation sont des cibles thérapeutiques importantes, et les KDACs dépendantes du zinc (appelées Histones Désacétylases, HDACs) ont été parmi les premières cibles des médicaments épigénétiques approuvés par la FDA (Food and Drug Administration) pour le traitement de certains cancers.
Notre équipe étudie les HDACs pour en comprendre les mécanismes enzymatiques mais aussi pour aider au développement de nouveaux candidats-médicaments qui peuvent cibler différents cancers ainsi que des maladies négligées (malaria, leishmaniose, schistosomiase, maladie de Chagas). Les sites actifs de chacune des HDACs montrent des spécificités propres mais aussi beaucoup de similitudes. Ainsi, à ce jour, de nombreuses questions restent en suspens sur la reconnaissance spécifique par les HDACs de leurs cibles protéiques et sur l’inhibition sélective des différentes isozymes des HDACs dans le but de développer des médicaments ayant un plus large spectre d’utilisation.
Notre équipe a initialement adressé ces problématiques en participant à deux larges projets européens (SEtTReND et A-ParaDDisE) qui ont réunis chercheurs européens, australiens et brésiliens pour développer des candidats-médicament ciblant sélectivement les HDACs de parasites pathogènes responsables de maladies négligées (malaria, leishmaniose, schistosomiase, maladie de Chagas). Nous avons pu ainsi résoudre la structure sous forme inhibée de deux HDACs pathogènes, la HDAC8 du ver parasite Schistosoma mansoni (smHDAC8), responsable de la schistosomiase, et la DAC2 du kinétoplastidé Trypanosoma cruzi (tcDAC2), responsable de la maladie de Chagas. Si la structure de smHDAC8 a montré une forte ressemblance à son homologue humain, certaines différences ont permis de progresser vers des inhibiteurs sélectifs pour l’enzyme pathogène.
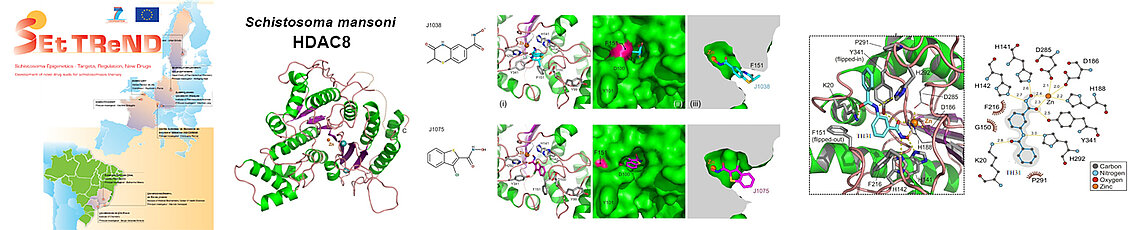
La HDAC8 du ver pathogène Schistosoma mansoni (smHDAC8) a été la cible majeure du projet européen SEtTReND (Schistosoma Epigenetics: Targets, Regulation, New Drugs). La résolution de la structure de cette HDAC a révélé ses spécificités structurales. Cela a permis de rechercher des premières molécules inhibitrices ayant une certaine sélectivité pour smHDAC8. Les structures des premiers inhibiteurs les plus prometteurs (J1038 et J1075) en complexe avec smHDAC8 ont montré leur mode de fixation à l'enzyme. Une étude d'optimisation de ces composés a ensuite été menée pour aboutir à une série de candidats-médicaments sélectifs de smHDAC8 par rapport aux HDACs humaines et de révéler les relations structure/fonction de cette enzyme.
La structure de tcDAC2 a par contre révélé des caractéristiques structurales très différentes des enzymes humaines, ce qui facilite le développement de candidats-médicaments sélectifs contre les trypanosomes, mais aussi contre les parasites responsables de la malaria et de la leishmaniose, ces pathogènes ayant aussi des HDACs atypiques mais dont les structures restent à déterminer. L’ensemble de nos études démontrent que les différences au niveau des sites actifs des HDACs, pathogènes mais aussi humaines, forment la base pour la reconnaissance spécifique de leurs cibles protéiques.
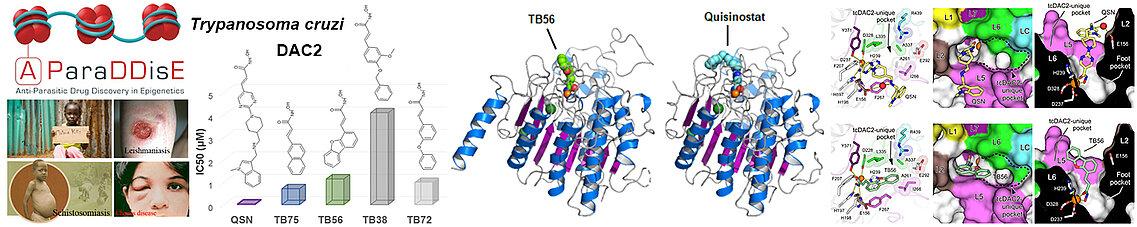
Le projet SEtTReND a apporté la preuve de concept de la possibilité de modifier des médicaments épigénétiques approuvés par la FDA pour en dériver des candidats-médicaments sélectifs contre les pathogènes eucaryotes. Le projet A-ParaDDisE (Anti-Parasitic Drug Discovery in Epigenetics) a appliqué cette stratégie aux enzymes épigénétiques d'autres pathogènes responsables de la malaria, de la leishmaniose et de la maladie de Chagas. Notamment, à travers notre étude intégrative de la DAC2 de Trypanosoma cruzi (tcDAC2), nous avons montré que les HDACs de ces pathogènes sont structuralement divergentes des HDACs humaines, ce qui va faciliter le développement de candidats-médicaments contre ces pathogènes.
Les objectifs de notre projet sur les désacétylases d'histones est de déchiffrer en termes biochimiques et structuraux les interactions spécifiques entre les HDACs et leurs protéines cibles, comprendre la dérégulation de leur activité dans les maladies, et de moduler ces interactions et ces activités à l’aide de petites molécules d’intérêt thérapeutique.
Publications sélectionnées
Marek et al. (2021) Species-selective targeting of pathogens revealed by the atypical structure and active site of Trypanosoma cruzi histone deacetylase DAC2. Cell Reports. DOI: 10.1016/j.celrep.2021.110129.
Ghazy et al. (2021) Synthesis, structure-activity relationships, cocrystallization and cellular characterization of novel smHDAC8 inhibitors for the treatment of schistosomiasis. Eur. J. Med. Chem., 225, 113745. doi: 10.1016/j.ejmech.2021.113745.
Kalinin et al. (2019) Structure-Based Design, Synthesis, and Biological Evaluation of Triazole-Based smHDAC8 Inhibitors. ChemMedChem, 15, 571-584. doi: 10.1002/cmdc.201900583.
Marek et al. (2018) Characterization of histone deacetylase 8 (HDAC8) selective inhibition reveals specific active site structural and functional determinants. J. Medicinal Chemistry. DOI: 10.1021/acs.jmedchem.8b01087.
Bayer et al. (2018) Synthesis, crystallization studies and in vitro characterization of novel cinnamic acid derivatives as SmHDAC8 inhibitors for the treatment of Schistosomiasis. ChemMedChem. DOI: 10.1002/cmdc.201800238.
Heimburg et al. (2017) Structure-Based Design and Biological Characterization of Selective Histone Deacetylase 8 (HDAC8) Inhibitors with Anti-Neuroblastoma Activity. J. Medicinal Chemistry. DOI: 10.1021/acs.jmedchem.7b01447.
Heimburg et al. (2016) Structure-based design and synthesis of potent inhibitors targeting HDAC8 of Schistosoma mansoni for the treatment of schistosomiasis. J. Medicinal Chemistry. DOI: 10.1021/acs.jmedchem.5b01478.
Chakrabarti et al. (2015) HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol. Sci., 36, 481-492. doi: 10.1016/j.tips.2015.04.013.
Marek et al. (2015) Drugging the schistosome zinc-dependent HDACs: current progress and future perspectives. Future Med Chem., 7, 783-800. doi: 10.4155/fmc.15.25.
Stolfa et al. (2014) Molecular basis for the antiparasitic activity of a mercaptoacetamide derivative that inhibits histone deacetylase 8 (HDAC8) from the human pathogen Schistosoma mansoni. J. Molecular Biology. DOI: 10.1016/j.jmb.2014.03.007.
Marek et al. (2013) Structural Basis for the Inhibition of Histone Deacetylase 8 (HDAC8), a Key Epigenetic Player in the Blood Fluke Schistosoma mansoni. PLoS Pathogens. DOI: 10.1371/journal.ppat.1003645.
Chaperons d’histones
Les protéines histones (H2A, H2B, H3, H4) forment l’échafaudage sur lequel se constituent les nucléosomes qui, eux-mêmes, forment l’unité de base de la chromatine. Parmi les histones, les histones dites canoniques sont notamment utilisées pour la formation de la chromatine lors de la division cellulaire, tandis que les histones dites variantes ont des rôles fonctionnels spécifiques à différents moments du cycle cellulaire et à différent endroits du génome. Les histones forment des paires spécifiques (H2A/H2B et H3/H4), et aucune de ces paires n’est trouvée libre, que ce soit dans le cytoplasme ou dans le noyau, pour éviter qu’elles puissent interagir de manière intempestive avec les acides nucléiques. Ainsi, les paires d’histones sont complexées à des chaperons d’histones jusqu’à ce qu’elles soient incorporées au sein de la chromatine.
Parmi les chaperons d’histones, certains ne sont spécifiques d’aucune paire d’histones en particulier. Certains autres chaperons sont spécifiques soit de la paire H2A/H2B soit de la paire H3/H4, sans distinction de leurs caractéristiques canoniques ou variantes. Enfin, d’autres chaperons vont reconnaitre précisément une paire canonique ou une paire variante spécifique. Les chaperons d’histones peuvent être impliqués dans le transport des paires d’histones entre les différents compartiments cellulaires ou au sein d’un même compartiment, tandis que d’autres vont être impliqués dans l’apport, l’échange et l’enlèvement des paires d’histones au sein de la chromatine. L’importance fonctionnelle des diverses paires d’histones et de leur intégration correcte au sein de la chromatine implique de comprendre leurs interactions avec leurs chaperons et comment ces derniers interagissent avec l’ensemble des effecteurs nucléaires.
Notre équipe adresse ces thématiques en étudiant principalement deux chaperons d’histones possédant des rôles différents : Spt6 et les chaperons du variant d’histone H2A.Z. Le chaperon Spt6 voyage avec l’ARN polymérase II. Spt6 reconstitue notamment les nucléosomes dans le sillage de la polymérase au cours de la transcription des gènes. Spt6 joue aussi un rôle de facteur d’élongation de la transcription et est couplé au système d’export des ARNs messagers. Nos études ont révélé que ce chaperon agit comme une plate-forme de recrutement de nombreux effecteurs nucléaires au cours des phases d’initiation et d’élongation de la transcription par l’ARN polymérase II, en interagissant avec ces effecteurs à l’aide de petits motifs spécifiques, lui permettant ainsi de jouer ses différents rôles fonctionnels.
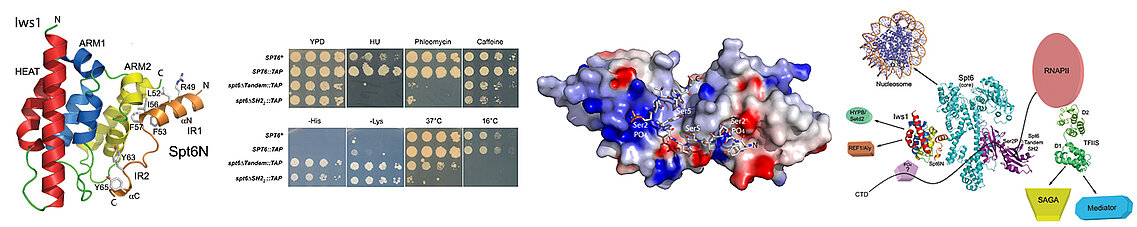
La protéine Spt6 possède à ses extrémités N- et C-terminales des motifs d'interaction avec différents effecteurs nucléaires. Nous avons résolu la structure d'un des motifs N-terminaux de Spt6 fixé à la protéine Iws1 qui est impliquée dans la maturation et l'export des ARN messagers. Nous avons aussi résolu la structure du tandem de motifs SH2 C-terminal de Spt6 qui interagit spécifiquement avec la queue C-terminale de l'ARN Polymérase II et qui permet à Spt6 de coopérer fonctionnellement avec cette polymérase. Nous avons valider nos données structurales et d'interaction avec des expériences de génétique de levure en collaboration avec l'équipe de Fred Winston à Harvard. Nos données démontrent du rôle de plateforme d'interaction et d'intégration de signaux joué par Spt6.
Dans le cas du variant d’histone H2A.Z, celui est reconnu par deux chaperons chez l’homme, YL1 et ANP32E. En collaboration avec l’équipe Hamiche de l’IGBMC, nous avons étudié la déposition et l’enlèvement de la paire H2A.Z/H2B au sein de la chromatine par ces deux chaperons. Nous avons ainsi mis en évidence les mécanismes similaires utilisés par ces deux chaperons pour reconnaître spécifiquement la paire H2A.Z/H2B. Par contre, nos travaux ont révélé les mécanismes divergents de ces deux chaperons pour s’assurer que la paire H2A.Z/H2B est correctement déposée au sein de la chromatine, dans le cas de YL1, et, est enlevée de la chromatine en reconnaissant un motif enfoui au cœur du nucléosome, dans le cas de ANP32E. Ces travaux ont aussi permis de mettre en évidence des liens fonctionnels entre ANP32E et le facteur de transcription/isolateur chromatinien CTCF.
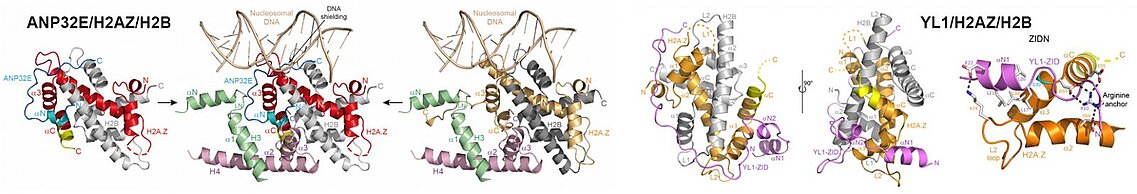
Le variant d'histone H2AZ joue des rôles fonctionnels spécifiques au cours du cycle cellulaire, étant notamment retrouvé dans les parties régulatrices du génome quand celles-ci sont impliquées dans la transcription des gènes. L'équipe Hamiche de l'IGBMC a mis en évidence deux chaperons de ce variant chez l'homme: ANP32E et YL1, la première enlevant la paire H2AZ/H2B de la chromatine, la seconde la déposant sur la chromatine. Nos structures de ces chaperons liés à la paire H2AZ/H2B ont montré qu'ils reconnaissaient tous deux le variant H2AZ du fait de l'absence d'un résidu glycine dans son hélice C-terminale et que leur interaction avec la paire H2AZ/H2B allongeait cette hélice C-terminale. Si ce changement de conformation de l'hélice C-terminale de H2AZ est suffisant à l'éviction de H2AZ/H2B de la chromatine par ANP32E, YL1 interagit de manière plus extensive et spécifique avec cette paire, s'assurant ainsi que c'est bien la paire H2AZ/H2B qui est déposée sur la chromatine.
Nous poursuivons ces travaux pour mieux comprendre comment les chaperons d’histones interagissent avec d’autres acteurs nucléaires, pour être dirigés spécifiquement vers certains domaines chromatiniens et pour recruter d’autres effecteurs épigénétiques.
Publications sélectionnées
Latrick, C.M. (2016) Molecular basis and specificity of H2A.Z–H2B recognition and deposition by the histone chaperone YL1. Nature Structural Molecular Biology. DOI: 10.1038/nsmb.3189.
Obri et al. (2014) ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature. DOI: 10.1038/nature12922.
Diebold et al. (2010) The structure of an Iws1/Spt6 complex reveals an interaction domain conserved in TFIIS, Elongin A and Med26. EMBO J. DOI: 10.1038/emboj.2010.272.
Diebold et al. (2010) Non-canonical tandem SH2 enables interaction of elongation factor Spt6 with RNA polymerase II. J. Biological Chemistry. DOI: 10.1074/jbc.M110.146696.
Complexes SMC
De nombreux processus nucléaires nécessitent le rapprochement spatial de différentes parties de notre génome ainsi que la formation de boucles de chromatine. Ces processus permettent de maintenir ensemble les chromatides sœurs lors de la réplication de l’ADN, puis leur condensation en chromosomes et leur répartition égale entre cellules filles au cours de la division cellulaire. Ces processus permettent aussi la réparation de l’ADN ainsi que la régulation de la transcription. L’ensemble de ces processus sont nécessaires tout au long du cycle cellulaire et dépendent chez les eucaryotes de complexes paralogues dits « Structural Maintenance of Chromosomes » (SMC). Chez les vertébrés, ceux-ci incluent les Cohésines mitotique et méiotique, les Condensines I et II, et le complexe SMC5/SMC6.
Les différents complexes SMC possèdent des organisations et des mécanismes similaires. Néanmoins, chacun possède des fonctions biologiques qui lui sont propres et qui sont assurées à l’aide de sous-unités cœurs et régulatrices spécifiques. Pour comprendre les fonctions de chacun des complexes SMC et leur perturbation dans les maladies, il est essentiel de pouvoir distinguer les mécanismes partagés par ces complexes et ceux qui leurs sont spécifiques. Notamment, l’ensemble des complexes SMC ont une activité ATPase essentielle à leurs fonctions portée par les têtes ATPases de chacune de leurs deux sous-unités cœur Smc. Comment ces sous-unités Smc fixent l’ATP et l’hydrolyse en ADP, et comment cette activité ATPase influence la conformation des complexes SMC, leur permettant ainsi de jouer leurs rôles fonctionnels, restent encore mal compris.
Notre équipe étudie la Cohésine mitotique humaine qui est essentielle pour la cohésion des chromatides sœurs, la séparation des chromosomes, la réparation de l’ADN, la régulation de la transcription, et la formation dynamique de boucles de chromatine et de domaines dits topologiquement associés (TADs). L’importance de la Cohésine est démontrée par sa mutation dans de très nombreux cancers liquides et solides et dans des pathologies appelées cohésinopathies qui sont des maladies neurodéveloppementales avec des traits autistiques. La Cohésine possède des protéines régulatrices qui lui sont spécifiques, y compris HDAC8 qui désacétyle la Cohésine au cours de son cycle fonctionnel, mais aussi CTCF qui interagit physiquement et fonctionnellement avec la Cohésine humaine pour former les boucles de chromatine et les TADs.
Notre équipe, en combinant des analyses biochimiques, biophysiques et structurales, par cristallographie et cryo-microscopie électronique, ainsi que in vivo, en utilisant le poisson-zèbre, a étudié l’activité ATPase de la Cohésine. Cette étude nous a permis de caractériser au niveau moléculaire l’ensemble des étapes du cycle ATPase de la Cohésine humaine, mettant en évidence des différences significatives entre les Cohésines vertébrées et celles de levures. De plus, nos travaux nous ont permis de comprendre la nécessité pour le complexe cœur de la Cohésine de s’associer à l’ADN et à sa sous-unité régulatrice NIPBL pour avoir une activité ATPase maximale, le complexe cœur n’ayant qu’une faible activité ATPase lorsqu’il seul ou associé à de l’ADN.
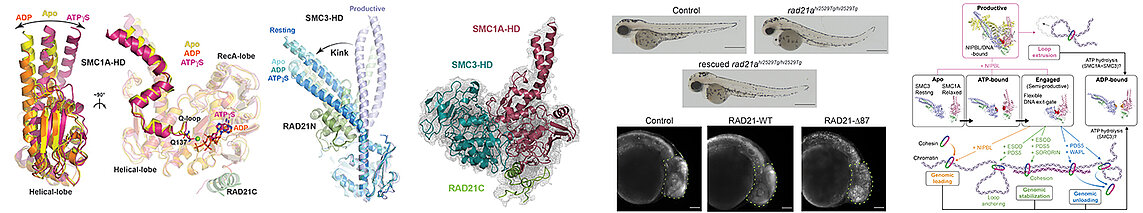
Le complexe Cohésine est un acteur majeur de la cohésion des chromatides sœurs, de la ségrégation des chromsomes et de l'organisation 3D du génome. La Cohésine agit grâce à des changements conformationnels qui sont induits par son activité de fixation et d'hydrolyse de l'ATP qui est modulée par différents régulateurs. La dérégulation de l'activité de la Cohésine par des mutations des ses sous-unités cœur et régulatrices aboutit à de très nombreux cancers et des maladies neurodévelopmentales appelées cohésinopathies. Pour mieux comprendre le mode d'action de la Cohésine humaine, nous avons caractérisé biochimiquement, biophysiquement et structuralement les mouvements de ses domaines ATPases, portés par ses sous-unités cœur SMC1A et SMC3, lorsque ceux-ci fixent ou hydrolyse l'ATP, et caractérisé l'interaction ATP-dépendante de ces domaines ATPases (panels de gauche). Nos différentes données nous permettent de décrire les changements conformationnels de la Cohésine au cours de son cycle ATPase (panel de droite). Nous avons utilisé le poisson-zèbre pour valider fonctionnellement in vivo nos données biochimisues et structurales (panel central).
Ces premiers travaux de notre équipe sur la Cohésine vertébrée ouvrent de larges perspectives pour étudier les mécanismes moléculaires qui régissent l’activité fonctionnelle de la Cohésine humaine, ses régulations, et son implication dans les cancers et les cohésinopathies.
Publications sélectionnées
Vitoria Gomes et al. (2022) Specific conformational dynamics of the ATPase head domains and DNA exit gate mediate the Cohesin ATPase cycle. BioRxiv, 2022.06.24.497451. doi: 10.1101/2022.06.24.497451.
Projets collaboratifs
Notre équipe a une longue tradition collaborative avec des équipes d’autres départements de l’IGBMC ainsi qu’en dehors de l’IGBMC. Nos premières collaborations nous ont permis d’aborder de nombreux sujets autour du cancer. Les nouvelles collaborations que nous avons établies nous amènent à étudier aussi les maladies neurodéveloppementales. Ces thématiques rejoignent directement nos questionnements scientifiques sur la Cohésine et son implication dans de très nombreux cancers et dans les cohésinopathies.
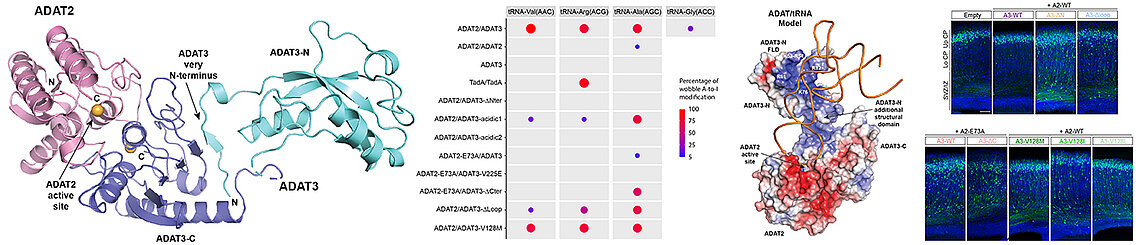
En collaboration avec les équipes de Juliette Godin à l'IGBMC et Laurence Drouard à l'IBMP, Strasbourg, nous avons caractérisé le complexe ADAT qui modifie les bases adenosine en position wobble des ARNt en inosine, ce qui permet d'étendre le code génétique. De plus, la mutation V128M dans le domaine N-terminal de sa sous-unité ADAT3 est responsable de déficiences intellectuelles et autres maladies neurodévelopmentales. Notre structure du complexe ADAT murin et nos données biophysiques a montré que le domaine N-terminal de ADAT3 permet de reconnaitre spécifiquement les ARNt pour les présenter au domaine catalytique de ADAT2. Ces données et nos essais enzymatiques ont montré que le domaine N-terminal de ADAT3 est essential à l'activité du complexe ADAT et que la mutation V128M gêne la présentation correcte de l'adenosine wobble à ADAT2 sans affecter cependant la fixation des ARNt.
Publications sélectionnées
Ramos-Morales et al. (2021) The structure of the mouse ADAT2/ADAT3 complex reveals the molecular basis for mammalian tRNA wobble adenosine-to-inosine deamination. Nucleic Acids Research. DOI: 10.1093/nar/gkab436.
Coassolo et al. (2021) Citrullination of pyruvate kinase M2 by PADI1 and PADI3 regulates glycolysis and cancer cell proliferation. Nature Communications. DOI: 10.1038/s41467-021-21960-4.
Alpern et al (2014) TAF4, a subunit of transcription factor II D, directs promoter occupancy of nuclear receptor HNF4A during post-natal hepatocyte differentiation. Elife. DOI: 10.7554/eLife.03613.
Nardini et al. (2013) NF-Y achieves CCAAT-specificity via a novel NF-YA DNA-reading module, and functions via an H2B-like modification. Cell. DOI: 10.1016/j.cell.2012.11.047.
Gangloff et al. (2001) The histone fold is a key structural motif of transcription factor TFIID. Trends Biochemical Sciences. DOI: 10.1016/s0968-0004(00)01741-2.
Ressources
Technologie de reconstitution de complexes protéiques
De nombreux effecteurs nucléaires, notamment ceux impliqués dans les mécanismes épigénétiques, agissent sous forme de complexes multiprotéiques qui sont difficiles à assembler et à purifier. De plus, la qualité et l’homogénéité des échantillons est primordiale pour les analyses in vitro, notamment structurales. Pour répondre à ces défis, notre équipe développe depuis plus de vingt ans une technologie de co-expression en bactérie Escherichia coli des sous-unités de complexes protéiques. Cette technologie permet de mettre en évidence, de reconstituer et de purifier des complexes protéiques, et est essentielle au développement de l’ensemble de nos projets. Cette technologie est cependant extrêmement versatile et peut s'appliquer à un grand nombre de projets scientifiques. Si vous voulez en savoir plus, vous trouverez ci-dessous un sous-ensemble de nos publications sur cette technologie. N'hésitez pas à nous contacter directement pour toute demande de renseignement, de matériel et/ou de collaboration.
Publications sélectionnées
Vincentelli & Romier (2016) Complex Reconstitution and Characterization by Combining Co-expression Techniques in Escherichia coli with High-Throughput. Adv. Exp. Med. Biol. DOI: 10.1007/978-3-319-27216-0_4.
Vincentelli & Romier (2013) Expression in Escherichia coli: becoming faster and more complex. Current Opinion Structural Biology. DOI: 10.1016/j.sbi.2013.01.006.
Diebold et al. (2011) Deciphering correct strategies for multiprotein complex assembly by co-expression: application to complexes as large as the histone octamer. J. Structural Biology. DOI: 10.1016/j.jsb.2011.02.001.
Romier et al. (2006) Co-expression of protein complexes in prokaryotic and eukaryotic hosts: experimental procedures, database tracking and case studies. Acta Cryst.D, 62, 1232-1242. doi: 10.1107/S0907444906031003.
Fribourg et al. (2001) Dissecting the interaction network of multiprotein complexes by pairwise coexpression of subunits in E. coli. J. Molecular Biology. DOI: 10.1006/jmbi.2000.4376.
Financements et partenaires











Collaborations et réseaux
Dans le but de développer nos projets dans une approche de biologie intégrative, notre équipe travaille de manière collaborative avec des équipes de recherche possédant des expertises complémentaires à la nôtre, en biologie mais aussi en chimie. De plus, notre technologie et notre savoir-faire quant à la reconstitution de complexes protéiques par multi-expression nous amène à developper des projets collaboratifs en plus de nos projets propres. Certaines de nos collaborations ont été établies depuis plus de deux décades, quand d'autres collaborations sont plus ponctuelles. Nos collaborations impliquent des équipes de l'IGBMC, des équipes strasbourgeoises, ainsi que des équipes au niveau national et international, comme démontré par nos publications.
Publications
2025
Article dans une revue
Disrupted transcriptional networks regulated by CHD1L during neurodevelopment underlie the mirrored neuroanatomical and growth phenotypes of the 1q21.1 copy number variant
- Marianne Victoria Lemée
- Maria Nicla Loviglio
- Tao Ye
- Peggy Tilly
- Céline Keime
- Chantal Weber
- Anastasiya Petrova
- Pernelle Klein
- Bastien Morlet
- Olivia Wendling
- Hugues Jacobs
- Mylène Tharreau
- David Geneviève
- Juliette Godin
- Christophe Romier
- Delphine Duteil
- Christelle Golzio
Nucleic Acids Research ; Volume: 53
Article dans une revue
ADAT3 variants disrupt the activity of the ADAT tRNA deaminase complex and impair neuronal migration
- Jordi Del-Pozo-Rodriguez
- Peggy Tilly
- Romain Lecat
- Hugo Rolando Vaca
- Laureline Mosser
- Elena Brivio
- Till Balla
- Marina Vitoria Gomes
- Elizabeth Ramos-Morales
- Noémie Schwaller
- Thalia Salinas-Giegé
- Grace Vannoy
- Eleina England
- Alysia Kern Lovgren
- Melanie O’leary
- Maya Chopra
- Naomi Meave Ojeda
- Mehran Beiraghi Toosi
- Atieh Eslahi
- Masoome Alerasool
- ...
Brain - A Journal of Neurology ; Volume: 148 ; Page: 3407–3421
2024
Article dans une revue
The cohesin ATPase cycle is mediated by specific conformational dynamics and interface plasticity of SMC1A and SMC3 ATPase domains
- Marina Vitoria Gomes
- Pauline Landwerlin
- Marie-Laure Diebold-Durand
- Tajith B Shaik
- Alexandre Durand
- Edouard Troesch
- Chantal Weber
- Karl Brillet
- Marianne V Lemée
- Christophe Decroos
- Ludivine Dulac
- Pierre Antony
- Erwan Watrin
- Eric Ennifar
- Christelle Golzio
- Christophe Romier
Cell Reports ; Volume: 43 ; Page: 114656
Article dans une revue
Structure–Activity Studies of 1,2,4-Oxadiazoles for the Inhibition of the NAD + -Dependent Lysine Deacylase Sirtuin 2
- Arianna Colcerasa
- Florian Friedrich
- Jelena Melesina
- Patrick Moser
- Anja Vogelmann
- Pavlos Tzortzoglou
- Emilia Neuwirt
- Manuela Sum
- Dina Robaa
- Lin Zhang
- Elizabeth Ramos-Morales
- Christophe Romier
- Oliver Einsle
- Eric Metzger
- Roland Schüle
- Olaf Groß
- Wolfgang Sippl
- Manfred Jung
Journal of Medicinal Chemistry ; Volume: 67 ; Page: 10076-10095
Article dans une revue
Integrated Experimental and Theoretical Investigation of Copper Active Site Properties of a Lytic Polysaccharide Monooxygenase from Serratia marcescens
- Alessia Munzone
- Manon Pujol
- Chris Joseph
- Ievgen Mazurenko
- Marius Réglier
- Sergio Jannuzzi
- Antoine Royant
- Giuseppe Sicoli
- Serena Debeer
- Maylis Orio
- A. Jalila Simaan
- Christophe Decroos
Inorganic Chemistry ; Volume: 63 ; Page: 11063–11078
Article dans une revue
Repurposing myoglobin into a carbene transferase for a [2,3]-sigmatropic Sommelet-Hauser rearrangement
- Manon Pujol
- Lison Degeilh
- Thibault Sauty de Chalon
- Marius Réglier
- A. Jalila Simaan
- Christophe Decroos
Journal of Inorganic Biochemistry ; Volume: 260 ; Page: 112688
2023
Article dans une revue
Development of First-in-Class Dual Sirt2/HDAC6 Inhibitors as Molecular Tools for Dual Inhibition of Tubulin Deacetylation
- Laura Sinatra
- Anja Vogelmann
- Florian Friedrich
- Margarita A. Tararina
- Emilia Neuwirt
- Arianna Colcerasa
- Philipp König
- Lara Toy
- Talha Z. Yesiloglu
- Sebastian Hilscher
- Lena Gaitzsch
- Niklas Papenkordt
- Shiyang Zhai
- Lin Zhang
- Christophe Romier
- Oliver Einsle
- Wolfgang Sippl
- Mike Schutkowski
- Olaf Gross
- Gerd Bendas
- ...
Journal of Medicinal Chemistry ; Volume: 66 ; Page: 14787-14814
Chapitre d’ouvrage
Synthesis, Biochemical, and Cellular Evaluation of HDAC6 Targeting Proteolysis Targeting Chimeras
- Salma Darwish
- Tino Heimburg
- Johannes Ridinger
- Daniel Herp
- Matthias Schmidt
- Christophe Romier
- Manfred Jung
- Ina Oehme
- Wolfgang Sippl
HDAC/HAT Function Assessment and Inhibitor Development ; Volume: 2589 ; Page: 179-193
Article dans une revue
LPMO‐like activity of bioinspired copper complexes: from model substrate to extended polysaccharides
- Rébecca Leblay
- Rafael Delgadillo-Ruiz
- Christophe Decroos
- Christelle Hureau
- Marius Réglier
- Ivan Castillo Pérez
- Bruno Faure
- A. Jalila Simaan
ChemCatChem ; Volume: 15 ; Page: e202300933
Article dans une revue
Chemically Diverse S. mansoni HDAC8 Inhibitors Reduce Viability in Worm Larval and Adult Stages
- Beatrice Noce
- Elisabetta Di Bello
- Clemens Zwergel
- Rossella Fioravanti
- Sergio Valente
- Dante Rotili
- Andrea Masotti
- Mohammad Salik Zeya Ansari
- Daniela Trisciuoglio
- Alokta Chakrabarti
- Christophe Romier
- Dina Robaa
- Wolfgang Sippl
- Manfred Jung
- Cécile Häberli
- Jennifer Keiser
- Antonello Mai
ChemMedChem ; Volume: 18
Page 1 sur 9
Biologie structurale intégrative -
Recherche contre le cancer -
Maladies rares