
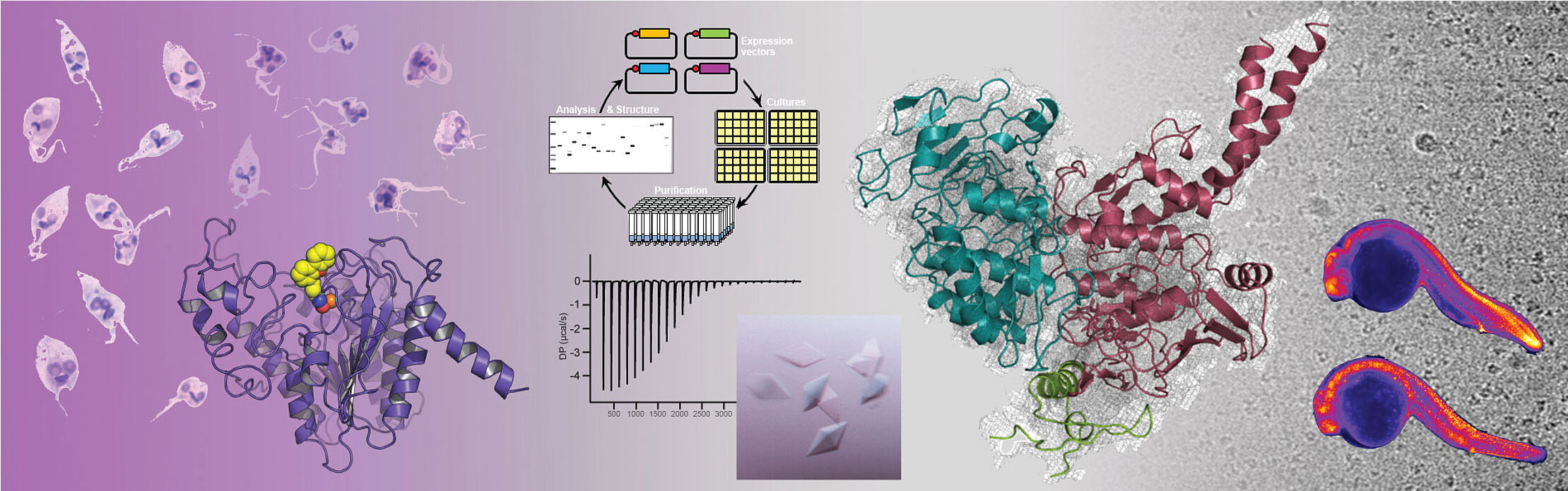
Molecular Basis of Chromatin and Transcription Regulation
Molecular Basis of Chromatin and Transcription Regulation

Our team studies epigenetic mechanisms to understand their involvement in nuclear processes, their dysfunctions in diseases, and how to modulate the activity of epigenetic effectors using drug candidates.
OUR INTERESTS
Understand the molecular bases of epigenetic mechanisms
Understand the regulation of nuclear processes by epigenetic effectors
Understand the dysregulation of epigenetic mechanisms in diseases
Understand how to modulate the activity of epigenetic effectors by drug candidates
Our team studies eukaryotic epigenetic mechanisms that are essential to cell homeostasis and the development of organisms. In particular, we characterize the molecular basis of epigenetic mechanisms to decipher their involvement in the regulation of nuclear processes and understand their physiological roles. We use this knowledge to determine how mutations in epigenetic effectors can lead to many different pathologies. We are also implementing strategies to characterize small molecules that inhibit or regulate the activity of epigenetic effectors, with the aim of helping the development of drug candidates targeting epigenetic pathologies, in particular cancers, neurodevelopmental and neglected diseases (malaria, leishmaniasis, schistosomiasis, Chagas disease).
OUR OBJECTIVES
Our objectives are to combine our different lines of research to characterize epigenetic mechanisms, understand their implications in pathologies, and participate in the development of epigenetic therapies. Our studies are based on in vitro biochemical, biophysical and structural biology analyses. However, we develop all of our projects within the framework of integrative biology strategies that combine in vitro, in cellulo and in vivo analyses. In particular, we are developing within the team the use of zebrafish to address our three research axes (mechanisms, pathologies and small molecules) in an animal model to characterize the structure/function relationships of our epigenetic targets.
OUR PROJECTS
Our scientific projects specifically target three types of epigenetic effectors: deacetylases, histone chaperones and SMC (Structural Maintenance of Chromosomes) complexes. Our epigenetic targets are all functionally related, our ultimate goal being to characterize their physical and functional interrelationships. In support of our epigenetic scientific projects, our team has been developing for more than two decades a technology for the reconstitution of protein complexes by multi-expression in bacterial hosts. This technology and our associated know-how are essential to all of our scientific projects and are also the source of specific collaborative projects with other research teams.
Members
Researchers
PhD students
Engineers
Technicians
Former members
PhD students
Pauline LANDWERLIN (2018-2022)
Marina VITORIA GOMES (2017-2021)
Elizabeth RAMOS MORALES (2017-2021)
Pernelle KLEIN (2016-2020)
Tajith Baba SHAIK (2014-2018)
Post-doctoral fellows
Nataliia ALEKSANDROVA (2016-2018)
Martin MAREK (2010-2017)
Régis BACK (2015-2017)
Michael KOCH (2007-2009)
Research subgroup(s)

Molecular mechanisms of chromatin reorganization
Subgroup Leader : Christophe ROMIER
Current projects
Histone deacetylases (HDAC)
Protein acetylation is a post-translational modification that regulates the activity of a very large number of cellular actors and that rivals phosphorylation in importance. In particular, acetylation plays an essential role in epigenetics by acting on chromatin compaction but also in epigenetic signaling. The acetylation mark is reversible, being deposited by lysine acetyl transferases (KATs), read by bromodomains, and removed by lysine deacetylases (KDACs). Enzymes of the acetylation cycle are important therapeutic targets, and zinc-dependent KDACs (called Histone Deacetylases, HDACs) were among the first targets of epigenetic drugs approved by the Food and Drug Administration (FDA) for the treatment of certain cancers.
Our team studies HDACs to understand their enzymatic mechanisms but also to help develop new drug candidates that can target different cancers as well as neglected diseases (malaria, leishmaniasis, schistosomiasis, Chagas disease). The active sites of each of the HDACs show many similarities but also own specificities. Thus, to date, many questions remain unanswered on the specific recognition by HDACs of their protein targets and on the selective inhibition of the different HDAC isozymes in order to develop drugs with a broader spectrum of use.
Our team initially addressed these issues by participating in two large European projects (SEtTReND and A-ParaDDisE) which brought together European, Australian and Brazilian researchers to develop drug candidates selectively targeting the HDACs of pathogenic parasites responsible for neglected diseases (malaria, leishmaniasis, schistosomiasis, Chagas disease). We were able to solve the structure in inhibited form of two pathogenic HDACs, HDAC8 of the parasitic worm Schistosoma mansoni (smHDAC8), responsible for schistosomiasis, and DAC2 of the kinetoplastid Trypanosoma cruzi (tcDAC2), responsible for Chagas disease. While the structure of smHDAC8 showed a strong resemblance to its human counterpart, certain differences allowed progress towards the development of selective inhibitors for this pathogenic enzyme.
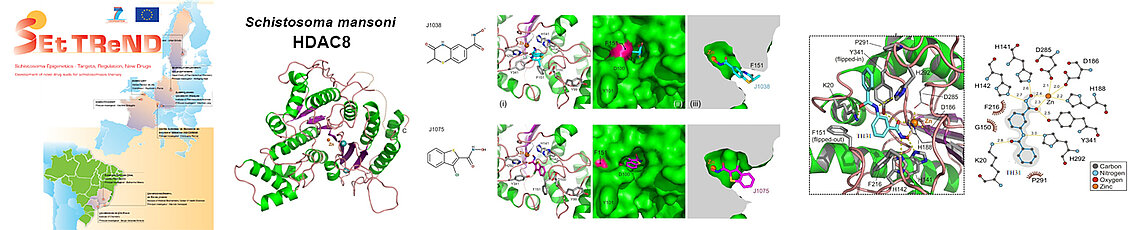
HDAC8 of the pathogenic worm Schistosoma mansoni (smHDAC8) was the major target of the European project SEtTReND (Schistosoma Epigenetics: Targets, Regulation, New Drugs). The resolution of the structure of this HDAC revealed its structural specificities. This made it possible to search for the first inhibitory molecules with a certain selectivity for smHDAC8. The structures of the first most promising inhibitors (J1038 and J1075) in complex with smHDAC8 showed their mode of binding to the enzyme. An optimization study of these compounds was then carried out to lead to a series of selective drug candidates for smHDAC8 compared to human HDACs and to reveal the structure/function relationships of this enzyme.
The structure of tcDAC2, on the other hand, revealed structural characteristics very different from human enzymes, which facilitates the development of selective drug candidates against trypanosomes, but also against the parasites responsible for malaria and leishmaniasis, these pathogens having also atypical HDACs but whose structures remain to be determined. All of our studies demonstrate that the differences in the active sites of HDACs, pathogenic but also human, form the basis for the specific recognition of their protein targets.
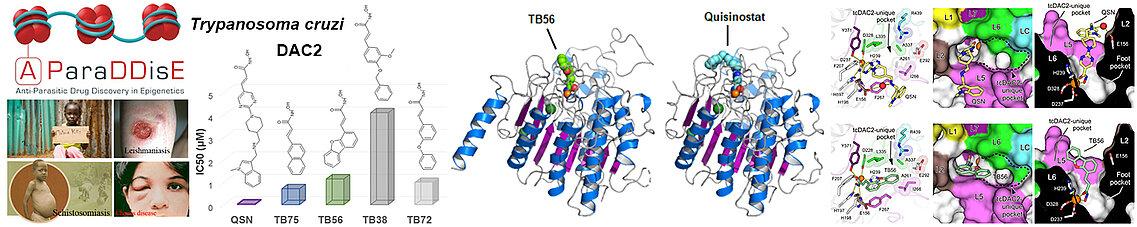
The SEtTReND project provided the proof of concept of the possibility of modifying epigenetic drugs approved by the FDA to derive selective drug candidates against eukaryotic pathogens. The A-ParaDDisE (Anti-Parasitic Drug Discovery in Epigenetics) project applied this strategy to the epigenetic enzymes of other pathogens responsible for malaria, leishmaniasis and the Chagas disease. In particular, through our integrative study of Trypanosoma cruzi DAC2 (tcDAC2), we have shown that the HDACs of these pathogens are structurally divergent from human HDACs, which will facilitate the development of drug candidates against these pathogens.
The objectives of our project on histone deacetylases is to decipher in biochemical and structural terms the specific interactions between HDACs and their target proteins, to understand the deregulation of their activity in diseases, and to modulate these interactions and these activities using small molecules of therapeutic interest.
Selected publications
Marek et al. (2021) Species-selective targeting of pathogens revealed by the atypical structure and active site of Trypanosoma cruzi histone deacetylase DAC2. Cell Reports. DOI: 10.1016/j.celrep.2021.110129.
Ghazy et al. (2021) Synthesis, structure-activity relationships, cocrystallization and cellular characterization of novel smHDAC8 inhibitors for the treatment of schistosomiasis. Eur. J. Med. Chem., 225, 113745. doi: 10.1016/j.ejmech.2021.113745.
Kalinin et al. (2019) Structure-Based Design, Synthesis, and Biological Evaluation of Triazole-Based smHDAC8 Inhibitors. ChemMedChem, 15, 571-584. doi: 10.1002/cmdc.201900583.
Marek et al. (2018) Characterization of histone deacetylase 8 (HDAC8) selective inhibition reveals specific active site structural and functional determinants. J. Medicinal Chemistry. DOI: 10.1021/acs.jmedchem.8b01087.
Bayer et al. (2018) Synthesis, crystallization studies and in vitro characterization of novel cinnamic acid derivatives as SmHDAC8 inhibitors for the treatment of Schistosomiasis. ChemMedChem. DOI: 10.1002/cmdc.201800238.
Heimburg et al. (2017) Structure-Based Design and Biological Characterization of Selective Histone Deacetylase 8 (HDAC8) Inhibitors with Anti-Neuroblastoma Activity. J. Medicinal Chemistry. DOI: 10.1021/acs.jmedchem.7b01447.
Heimburg et al. (2016) Structure-based design and synthesis of potent inhibitors targeting HDAC8 of Schistosoma mansoni for the treatment of schistosomiasis. J. Medicinal Chemistry. DOI: 10.1021/acs.jmedchem.5b01478.
Chakrabarti et al. (2015) HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol. Sci., 36, 481-492. doi: 10.1016/j.tips.2015.04.013.
Marek et al. (2015) Drugging the schistosome zinc-dependent HDACs: current progress and future perspectives. Future Med Chem., 7, 783-800. doi: 10.4155/fmc.15.25.
Stolfa et al. (2014) Molecular basis for the antiparasitic activity of a mercaptoacetamide derivative that inhibits histone deacetylase 8 (HDAC8) from the human pathogen Schistosoma mansoni. J. Molecular Biology. DOI: 10.1016/j.jmb.2014.03.007.
Marek et al. (2013) Structural Basis for the Inhibition of Histone Deacetylase 8 (HDAC8), a Key Epigenetic Player in the Blood Fluke Schistosoma mansoni. PLoS Pathogens. DOI: 10.1371/journal.ppat.1003645.
Histone chaperones
The histone proteins (H2A, H2B, H3, H4) form the scaffolding on which the nucleosomes are assembled, which themselves form the basic unit of chromatin. Among the histones, the canonical histones are used in particular for the formation of chromatin during cell division, while the histone variants have specific functional roles at different times of the cell cycle and at different locations in the genome. The histones form specific pairs (H2A/H2B and H3/H4), and none of these pairs is found free, either in the cytoplasm or in the nucleus, to prevent them from being able to interact in an inopportune manner with the nucleic acids. Thus, histone pairs are complexed to histone chaperones until they are incorporated into chromatin.
Among histone chaperones, some are not specific to any particular histone pair. Some other chaperones are specific to either the H2A/H2B pair or the H3/H4 pair, regardless of their canonical or variant characteristics. Finally, other chaperones will precisely recognize a canonical pair or a specific variant pair. Histone chaperones can be involved in the transport of histone pairs between different cellular compartments or within the same compartment, while others will be involved in supply, exchange and removal of the histone pairs within chromatin. The functional importance of the various histone pairs and their correct integration within the chromatin implies understanding their interactions with their chaperones and how the latter interact with the nuclear effectors.
Our team addresses these themes by studying mainly two histone chaperones with different roles: Spt6 and the chaperones of the histone variant H2A.Z. The Spt6 chaperone travels with RNA polymerase II. Spt6 notably reconstitutes nucleosomes in the wake of this polymerase during gene transcription. Spt6 also acts as a transcription elongation factor and is coupled to the messenger RNA export system. Our studies revealed that this chaperone acts as a recruitment platform for numerous nuclear effectors during the initiation and elongation phases of transcription by RNA polymerase II. Notably, Spt6 to play its various functional roles by interacting with these nuclear effectors by interacting with them using short specific patterns.
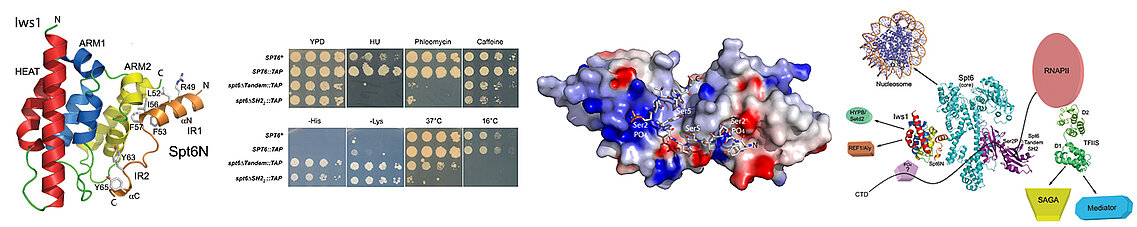
The N- and C-termini of the Spt6 protein has, at its N- and C-terminal harbour specific motifs of interaction with various nuclear effectors. We have resolved the structure of one of the N-terminal motifs of Spt6 attached to the Iws1 protein, which is involved in the maturation and export of messenger RNAs. We have also solved the structure of the C-terminal tandem of SH2 motifs of Spt6, which interacts specifically with the C-terminal tail of RNA Polymerase II and which allows Spt6 to cooperate functionally with this polymerase. We validated our structural and interaction data with yeast genetic experiments in collaboration with Fred Winston's team at Harvard. Our data demonstrate the role of interaction and signal integration platform played by Spt6.
In the case of the histone variant H2A.Z, it is recognized by two chaperones in humans, YL1 and ANP32E. In collaboration with the Hamiche team of the IGBMC, we studied the deposition and removal of the H2A.Z/H2B pair within the chromatin by these two chaperones. We have highlighted the similar mechanisms used by these two chaperones to specifically recognize the H2A.Z/H2B pair. On the other hand, our work has revealed the divergent mechanisms of these two chaperones to ensure that the H2A.Z/H2B pair is correctly deposited within the chromatin, in the case of YL1, and is removed from the chromatin by recognizing a motif buried in the heart of the nucleosome, in the case of ANP32E. This work also revealed functional links between ANP32E and the transcription factor/chromatin insulator CTCF.
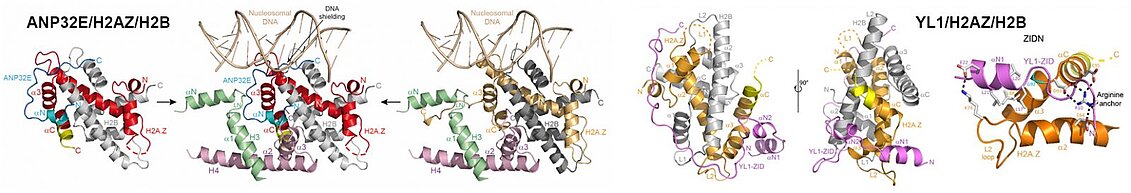
The H2AZ histone variant plays specific functional roles during the cell cycle, being found in particular in the regulatory parts of the genome when these are involved in gene transcription. The Hamiche team from the IGBMC has found two chaperones of this variant in humans: ANP32E and YL1, the first removing the H2AZ/H2B pair from chromatin, the second depositing it on the chromatin. Our structures of these chaperones linked to the H2AZ/H2B pair showed that they both recognize the H2AZ variant due to the absence of a glycine residue in its C-terminal helix and that their interaction with the H2AZ/H2B pair lengthened this C-terminal helix. If this conformational change of the C-terminal helix of H2AZ is sufficient for the eviction of H2AZ/H2B from chromatin by ANP32E, YL1 interacts more extensively and specifically with this pair, thus ensuring that it is the H2AZ/H2B pair which is correctly deposited on the chromatin.
We are pursuing this work to better understand how histone chaperones interact with other nuclear actors, to be directed specifically to certain chromatin domains and to recruit other epigenetic effectors.
Selected publications
Latrick, C.M. (2016) Molecular basis and specificity of H2A.Z–H2B recognition and deposition by the histone chaperone YL1. Nature Structural Molecular Biology. DOI: 10.1038/nsmb.3189.
Obri et al. (2014) ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature. DOI: 10.1038/nature12922.
Diebold et al. (2010) The structure of an Iws1/Spt6 complex reveals an interaction domain conserved in TFIIS, Elongin A and Med26. EMBO J. DOI: 10.1038/emboj.2010.272.
Diebold et al. (2010) Non-canonical tandem SH2 enables interaction of elongation factor Spt6 with RNA polymerase II. J. Biological Chemistry. DOI: 10.1074/jbc.M110.146696.
SMC complexes
Many nuclear processes require the spatial rapprochement of different parts of our genome as well as the formation of chromatin loops. These processes keep sister chromatids together during DNA replication, then condense them into chromosomes and distribute them evenly among daughter cells during cell division. These processes also allow DNA repair and the regulation of transcription. All of these processes are required throughout the cell cycle and depend in eukaryotes on paralogous complexes called “Structural Maintenance of Chromosomes” (SMC). In vertebrates, these include mitotic and meiotic Cohesins, Condensins I and II, and the SMC5/SMC6 complex.
The different SMC complexes have similar organizations and mechanisms. However, each has its own biological functions which are ensured with the help of specific core and regulatory subunits. To understand the functions of each of the SMC complexes and their dysregulation in diseases, it is essential to be able to distinguish the mechanisms shared by these complexes and those which are specific to them. In particular, all of the SMC complexes have an ATPase activity essential to their functions carried by the ATPase heads of each of their two core Smc subunits. How these Smc subunits bind ATP and hydrolyze it to ADP, and how this ATPase activity influences the conformation of SMC complexes, thus allowing them to perform their functional roles, remain poorly understood.
Our team studies human mitotic cohesin which is essential for sister chromatid cohesion, chromosome separation, DNA repair, regulation of transcription, and for the dynamic formation of chromatin loops and topologically associated domains (TADs). The importance of Cohesin is demonstrated by its mutation in many liquid and solid cancers and in pathologies called cohesinopathies which are neurodevelopmental diseases with autistic traits. Cohesin has specific regulatory proteins, including HDAC8 which deacetylates Cohesin during its functional cycle, but also CTCF which physically and functionally interacts with human Cohesin to form chromatin loops and TADs.
Our team studied the ATPase activity of Cohesin by combining biochemical, biophysical and structural, by crystallography and cryo-electron microscopy, in vitro analyses as well as in vivo, using zebrafish. This study allowed us to characterize at the molecular level the different stages of the ATPase cycle of human Cohesin, highlighting significant differences between vertebrate Cohesins and those of yeasts. In addition, our work has enabled us to understand the requirement for the core Cohesin complex to associate with DNA and the regulatory subunit NIPBL to have a maximum ATPase activity, the core complex having only low ATPase activity when alone or associated with DNA.
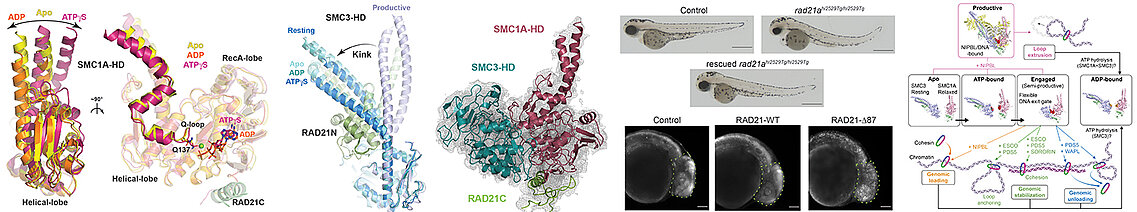
The Cohesin complex is a major player in sister chromatid cohesion, chromsome segregation and 3D genome organization . Cohesin acts through conformational changes which are induced by its ATP binding and hydrolysis activity which is modulated by different regulators. The deregulation of the activity of Cohesin by mutations of its core and regulatory subunits leads to a large number of cancers and neurodevelopmental diseases called cohesinopathies. To better understand the mode of action of human Cohesin, we characterized biochemically, biophysically and structurally the movements of its ATPase domains, carried by its core subunits SMC1A and SMC3, when these bind or hydrolyze ATP, and characterized the ATP-dependent interaction of these ATPase domains (left panels). Our different data allow us to describe the conformational changes of Cohesin during its ATPase cycle (right panel). We used zebrafish to functionally validate in vivo our biochemical and structural data (central panel).
This initial work by our team on vertebrate Cohesin opens up broad perspectives for studying the molecular mechanisms that govern the functional activity of human Cohesin, its regulations, and its involvement in cancers and cohesinopathies.
Selected publications
Vitoria Gomes et al. (2022) Specific conformational dynamics of the ATPase head domains and DNA exit gate mediate the Cohesin ATPase cycle. BioRxiv, 2022.06.24.497451. doi: 10.1101/2022.06.24.497451.
Collaborative projects
Our team has a long tradition of collaborating with teams from other departments within the IGBMC as well as outside of the IGBMC. Our first collaborations allowed us to address many topics around cancer. The new collaborations that we have established lead us to also study neurodevelopmental diseases. These themes are directly related to our scientific questions on Cohesin and its involvement in many cancers and in cohesinopathies.
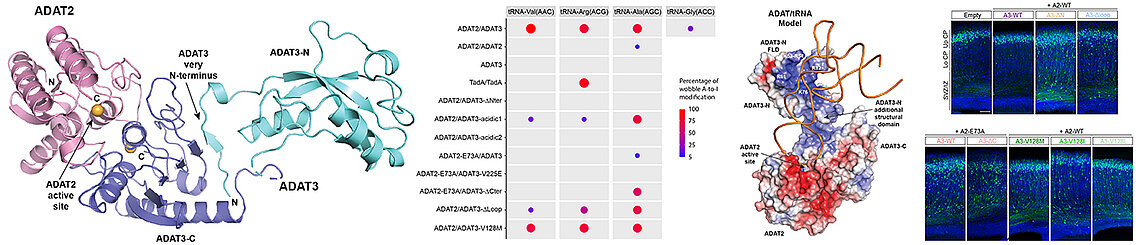
In collaboration with the teams of Juliette Godin at the IGBMC and Laurence Drouard at the IBMP, Strasbourg, we have characterized the ADAT complex which modifies the adenosine bases at the wobble position of tRNAs into inosine, which enables the extension of the genetic code genetic. In addition, the V128M mutation in the N-terminal domain of its ADAT3 subunit is responsible for intellectual disabilities and other neurodevelopmental diseases. Our structure of the murine ADAT complex and our biophysical data showed that the N-terminal domain of ADAT3 enables the specific recognition of tRNAs to present them to the catalytic domain of ADAT2. These data and our enzymatic assays showed that the N-terminal domain of ADAT3 is essential for the activity of the ADAT complex and that the V128M mutation interferes with the correct presentation of the adenosine wobble to ADAT2 without however affecting tRNA binding.
Selected publications
Ramos-Morales et al. (2021) The structure of the mouse ADAT2/ADAT3 complex reveals the molecular basis for mammalian tRNA wobble adenosine-to-inosine deamination. Nucleic Acids Research. DOI: 10.1093/nar/gkab436.
Coassolo et al. (2021) Citrullination of pyruvate kinase M2 by PADI1 and PADI3 regulates glycolysis and cancer cell proliferation. Nature Communications. DOI: 10.1038/s41467-021-21960-4.
Alpern et al (2014) TAF4, a subunit of transcription factor II D, directs promoter occupancy of nuclear receptor HNF4A during post-natal hepatocyte differentiation. Elife. DOI: 10.7554/eLife.03613.
Nardini et al. (2013) NF-Y achieves CCAAT-specificity via a novel NF-YA DNA-reading module, and functions via an H2B-like modification. Cell. DOI: 10.1016/j.cell.2012.11.047.
- Gangloff et al. (2001) The histone fold is a key structural motif of transcription factor TFIID. Trends Biochemical Sciences. DOI: 10.1016/s0968-0004(00)01741-2.
Resources
Protein complexes reconstitution technology
Many nuclear effectors, especially those involved in epigenetic mechanisms, act as multiprotein complexes that are difficult to assemble and purify. In addition, the quality and homogeneity of the samples is essential for in vitro analyses, particularly structural ones. To meet these challenges, our team has been developing for more than twenty years a technology for the co-expression in the Escherichia coli bacteria of protein complex subunits. This technology makes it possible to characterize, reconstitute and purify protein complexes, and is essential to the development of all of our projects. This technology is however extremely versatile and can be applied to a large number of scientific projects. If you want to learn more about it, find below a subset of our publications on this technology. Also, do not hesitate to contact us directly for any request for information, material and/or collaboration.
Selected publications
Vincentelli & Romier (2016) Complex Reconstitution and Characterization by Combining Co-expression Techniques in Escherichia coli with High-Throughput. Adv. Exp. Med. Biol. DOI: 10.1007/978-3-319-27216-0_4.
Vincentelli & Romier (2013) Expression in Escherichia coli: becoming faster and more complex. Current Opinion Structural Biology. DOI: 10.1016/j.sbi.2013.01.006.
Diebold et al. (2011) Deciphering correct strategies for multiprotein complex assembly by co-expression: application to complexes as large as the histone octamer. J. Structural Biology. DOI: 10.1016/j.jsb.2011.02.001.
Romier et al. (2006) Co-expression of protein complexes in prokaryotic and eukaryotic hosts: experimental procedures, database tracking and case studies. Acta Cryst.D, 62, 1232-1242. doi: 10.1107/S0907444906031003.
- Fribourg et al. (2001) Dissecting the interaction network of multiprotein complexes by pairwise coexpression of subunits in E. coli. J. Molecular Biology. DOI: 10.1006/jmbi.2000.4376.
Collaborations and networks
In order to develop our projects in an integrative biology approach, our team works collaboratively with research teams with complementary expertise to ours, in biology but also in chemistry. In addition, our technology and our know-how in the reconstitution of protein complexes by multi-expression leads us to develop collaborative projects in addition to our own projects. Some of our collaborations have been established for more than two decades, while other collaborations are more occasional. Our collaborations involve teams from the IGBMC, teams from Strasbourg, as well as teams at the national and international levels, as demonstrated by our publications.
Funding and partners











Publications
2025
Article in a journal
Disrupted transcriptional networks regulated by CHD1L during neurodevelopment underlie the mirrored neuroanatomical and growth phenotypes of the 1q21.1 copy number variant
- Marianne Victoria Lemée
- Maria Nicla Loviglio
- Tao Ye
- Peggy Tilly
- Céline Keime
- Chantal Weber
- Anastasiya Petrova
- Pernelle Klein
- Bastien Morlet
- Olivia Wendling
- Hugues Jacobs
- Mylène Tharreau
- David Geneviève
- Juliette Godin
- Christophe Romier
- Delphine Duteil
- Christelle Golzio
Nucleic Acids Research ; Volume: 53
Article in a journal
ADAT3 variants disrupt the activity of the ADAT tRNA deaminase complex and impair neuronal migration
- Jordi Del-Pozo-Rodriguez
- Peggy Tilly
- Romain Lecat
- Hugo Rolando Vaca
- Laureline Mosser
- Elena Brivio
- Till Balla
- Marina Vitoria Gomes
- Elizabeth Ramos-Morales
- Noémie Schwaller
- Thalia Salinas-Giegé
- Grace Vannoy
- Eleina England
- Alysia Kern Lovgren
- Melanie O’leary
- Maya Chopra
- Naomi Meave Ojeda
- Mehran Beiraghi Toosi
- Atieh Eslahi
- Masoome Alerasool
- ...
Brain - A Journal of Neurology ; Volume: 148 ; Page: 3407–3421
2024
Article in a journal
The cohesin ATPase cycle is mediated by specific conformational dynamics and interface plasticity of SMC1A and SMC3 ATPase domains
- Marina Vitoria Gomes
- Pauline Landwerlin
- Marie-Laure Diebold-Durand
- Tajith B Shaik
- Alexandre Durand
- Edouard Troesch
- Chantal Weber
- Karl Brillet
- Marianne V Lemée
- Christophe Decroos
- Ludivine Dulac
- Pierre Antony
- Erwan Watrin
- Eric Ennifar
- Christelle Golzio
- Christophe Romier
Cell Reports ; Volume: 43 ; Page: 114656
Article in a journal
Structure–Activity Studies of 1,2,4-Oxadiazoles for the Inhibition of the NAD + -Dependent Lysine Deacylase Sirtuin 2
- Arianna Colcerasa
- Florian Friedrich
- Jelena Melesina
- Patrick Moser
- Anja Vogelmann
- Pavlos Tzortzoglou
- Emilia Neuwirt
- Manuela Sum
- Dina Robaa
- Lin Zhang
- Elizabeth Ramos-Morales
- Christophe Romier
- Oliver Einsle
- Eric Metzger
- Roland Schüle
- Olaf Groß
- Wolfgang Sippl
- Manfred Jung
Journal of Medicinal Chemistry ; Volume: 67 ; Page: 10076-10095
Article in a journal
Integrated Experimental and Theoretical Investigation of Copper Active Site Properties of a Lytic Polysaccharide Monooxygenase from Serratia marcescens
- Alessia Munzone
- Manon Pujol
- Chris Joseph
- Ievgen Mazurenko
- Marius Réglier
- Sergio Jannuzzi
- Antoine Royant
- Giuseppe Sicoli
- Serena Debeer
- Maylis Orio
- A. Jalila Simaan
- Christophe Decroos
Inorganic Chemistry ; Volume: 63 ; Page: 11063–11078
Article in a journal
Repurposing myoglobin into a carbene transferase for a [2,3]-sigmatropic Sommelet-Hauser rearrangement
- Manon Pujol
- Lison Degeilh
- Thibault Sauty de Chalon
- Marius Réglier
- A. Jalila Simaan
- Christophe Decroos
Journal of Inorganic Biochemistry ; Volume: 260 ; Page: 112688
2023
Article in a journal
Development of First-in-Class Dual Sirt2/HDAC6 Inhibitors as Molecular Tools for Dual Inhibition of Tubulin Deacetylation
- Laura Sinatra
- Anja Vogelmann
- Florian Friedrich
- Margarita A. Tararina
- Emilia Neuwirt
- Arianna Colcerasa
- Philipp König
- Lara Toy
- Talha Z. Yesiloglu
- Sebastian Hilscher
- Lena Gaitzsch
- Niklas Papenkordt
- Shiyang Zhai
- Lin Zhang
- Christophe Romier
- Oliver Einsle
- Wolfgang Sippl
- Mike Schutkowski
- Olaf Gross
- Gerd Bendas
- ...
Journal of Medicinal Chemistry ; Volume: 66 ; Page: 14787-14814
Chapter of the book
Synthesis, Biochemical, and Cellular Evaluation of HDAC6 Targeting Proteolysis Targeting Chimeras
- Salma Darwish
- Tino Heimburg
- Johannes Ridinger
- Daniel Herp
- Matthias Schmidt
- Christophe Romier
- Manfred Jung
- Ina Oehme
- Wolfgang Sippl
HDAC/HAT Function Assessment and Inhibitor Development ; Volume: 2589 ; Page: 179-193
Article in a journal
LPMO‐like activity of bioinspired copper complexes: from model substrate to extended polysaccharides
- Rébecca Leblay
- Rafael Delgadillo-Ruiz
- Christophe Decroos
- Christelle Hureau
- Marius Réglier
- Ivan Castillo Pérez
- Bruno Faure
- A. Jalila Simaan
ChemCatChem ; Volume: 15 ; Page: e202300933
Article in a journal
Chemically Diverse S. mansoni HDAC8 Inhibitors Reduce Viability in Worm Larval and Adult Stages
- Beatrice Noce
- Elisabetta Di Bello
- Clemens Zwergel
- Rossella Fioravanti
- Sergio Valente
- Dante Rotili
- Andrea Masotti
- Mohammad Salik Zeya Ansari
- Daniela Trisciuoglio
- Alokta Chakrabarti
- Christophe Romier
- Dina Robaa
- Wolfgang Sippl
- Manfred Jung
- Cécile Häberli
- Jennifer Keiser
- Antonello Mai
ChemMedChem ; Volume: 18
Integrated structural biology -
Cancer research -
Rare diseases