
Maladies à gain de fonction d'ARN
Maladies à gain de fonction d'ARN
Les microsatellites sont des séquences d’ADN de 3 à 6 nucléotides répétés de multiples fois dans le génome. Des expansions anormales de ces microsatellites peuvent conduire à une quarantaine de maladies génétiques. La thématique de notre groupe est de mieux comprendre comment de telles expansions de répétitions, mais situées dans les régions dites « non codantes » du génome (5’UTR, Introns, 3’UTR, etc.), peuvent conduire à des atteintes neuromusculaires et/ ou neurodégénératives. Nous étudions principalement les pathologies suivantes :
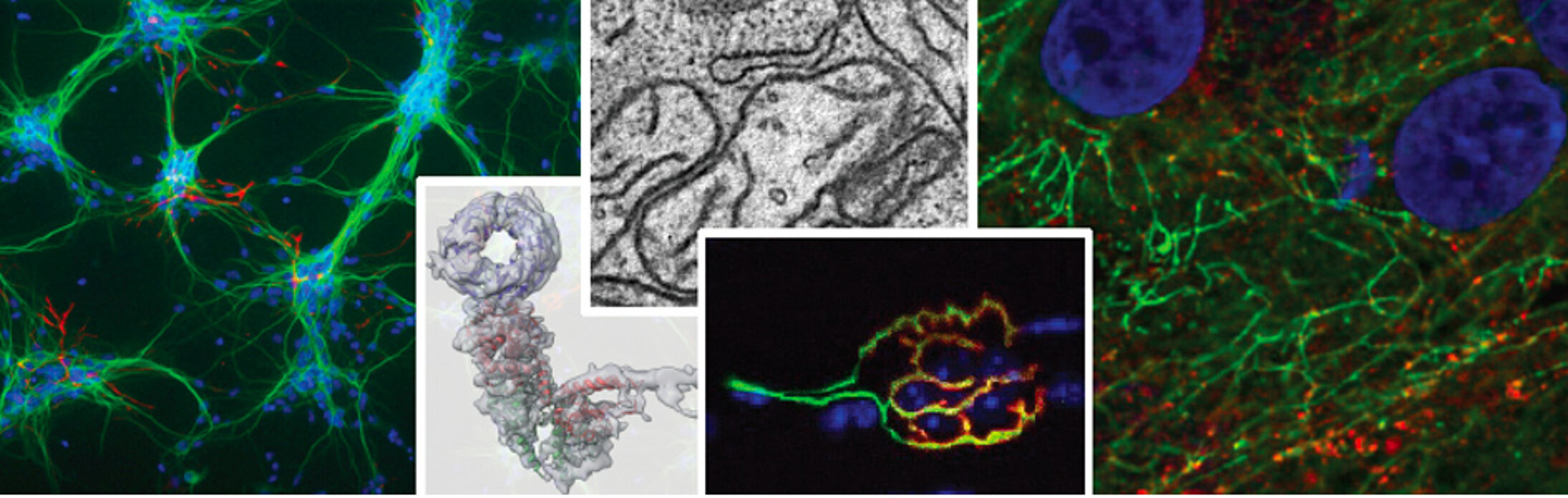
- Les dystrophies myotonique (DM) sont les dystrophies les plus communes chez l’adulte et sont dues respectivement à des expansions de répétitions CTG (DM1) ou CCTG (DM2) dans le 3’UTR du gène DMPK ou le premier intron du gène CNBP. Ces répétitions sont transcrites en ARN CUG ou CCUG qui lient et séquestrent les protéines MBNL, des facteurs d’épissage, conduisant ainsi à des altérations spécifiques de l’épissage alternatif causant les symptômes de ces maladies. Nos travaux sur les dystrophies myotonique comprennent : Fugier et al., Nature Medicine, 2011 ; Rau et al., Nature Structural and Molecular Biology, 2011 ; Freyermuth et al., Nature Communication, 2016 ; Sellier et al., Nature Communication, 2018 ; etc.
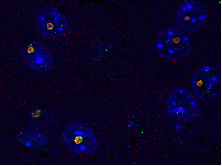
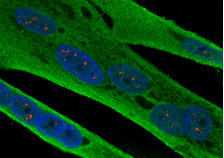
- Le syndrome de tremblements et d’ataxie associé à l’X fragile (FXTAS) et la maladie à inclusion intranucléaire neuronale (NIID) sont des maladies neurodégénératives causées par des expansions de répétitions GGC respectivement situées dans le 5’UTR des gène FMR1 et NOTCH2NLC. Nos données montrent que ces répétitions sont traduites dans des protéines polyglycines qui forment des inclusions cellulaires et sont toxiques en modèle cellulaires et animaux (Sellier et al., Cell reports, 2013 ; Sellier et al., Neuron, 2017 ; Boivin et al., Neuron, 2021 ; etc.).
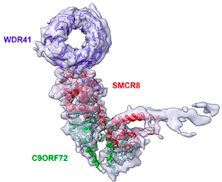
- La sclérose latérale amyotrophique associée à une démence fronto-temporale (SLA-DFT) est la troisième maladie neurodégénérative la plus commune après les maladies d’Alzheimer et de Parkinson. La SLA ou maladie de Charcot est due à la mort des motoneurones entrainant une paralysie progressive des muscles impliqués dans la motricité volontaire, conduisant à la mort des patients après 3 à 5 ans d'évolution. La cause la plus fréquente de SLA-DFT est une expansion de répétitions GGGGCC (G4C2) dans le premier intron du gène C9ORF72. Ces répétitions G4C2 sont traduites en protéines toxiques. De plus, ces répétitions conduisent à des modifications épigénétiques de l’ADN diminuant l’expression du gène et donc de la protéine C9ORF72. Nous avons montré que cette protéine appartient à un complexe protéique qui régule de petites protéines GTPases impliquées dans le trafic vésiculaire, notamment l’autophagie, un mécanisme de dégradation essentiel pour les neurones (Sellier et al., EMBO Journal, 2016 ; Boivin et al., EMBO Journal, 2020 ; etc.).
Notre but est de
Découvrir par quel mécanismes moléculaires ces répétitions sont pathogéniques,
Établir des modèles cellulaires (iPS, cultures primaires de cellules musculaires et de neurones, etc.) et animaux (souris) pertinent pour étudier ces maladies
Identifier des molécules pharmacologiques rétablissant un fonctionnement normal dans ces modèles cellulaires et animaux.
Pour cela, nous développons et utilisons une variété d’approches et de techniques allant de la biochimie et biologie moléculaire (protéines recombinantes, purification de complexe protéiques ou ARN-protéines, etc.), biologie cellulaires (lentivirus, AAV, iPS, motoneurones, culture primaire de neurones ou de cellule musculaires, etc.), approches à large échelles (transcriptomique, protéomique, etc.) et pathophysiologie de modèles animaux (souris transgénique et knockout).
En conclusion, notre travail a pour but de clarifier par quels mécanismes moléculaires et cellulaires des expansions de répétitions conduisent à des dysfonctionnements et à la mort des neurones et des cellules musculaires, afin d’identifier des approches thérapeutiques pour ces maladies dévastatrices.
![]()
Membres
Chercheur(euse)s
Doctorant(e)s
Ingénieur(eure)s
Anciens membres
Charlotte FUGIER (Former Engineer)
Angeline GAUCHEROT (Former Engineer)
Frédérique RAU (Former PhD student)
Fernande FREYERMUTH (Former PhD student)
Michel NEY (Former PhD student)
Camille CORBIER (Former PhD student)
Projets en cours
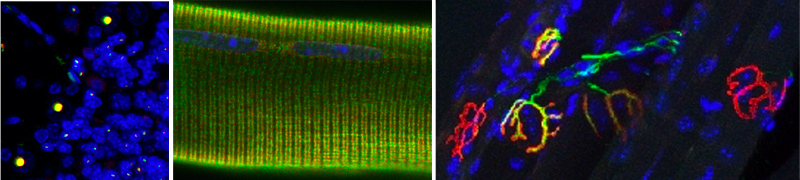
Nous avons plusieurs nouveaux projets en cours ou en développement :
- Nous étudions de nouvelles expansions de répétitions transcrites en ARN « toxiques » liant et titrant des protéines spécifiques dans des maladies neurodégénératives. Ce mécanisme est similaire à la titration des facteurs d’épissage MBNL par les répétitions ARN CUG conduisant à des défauts d’épissages spécifiques dans les dystrophies myotoniques (Fugier et al., Nature Medicine 2011 ; Freyermuth et al., Nature Communication 2016 ; Sellier et al., Nature Communication 2018).
- Nous nous intéressons à de nouvelles expansions de répétitions GGC potentiellement traduites en de nouvelles protéines polyGlycines toxiques dans des maladies génétiques neuromusculaires et/ ou neurodégénératives. Ce projet est une extension directe de notre travail sur FXTAS and NIID (Sellier et al., Neuron 2017 ; Boivin et al., Neuron 2021).
- Nous avons identifié de nouvelles protéines interagissant avec le complexe C9ORF72 et poursuivons l’étude de la fonction cellulaire, de la composition mais aussi de la structure par cryoEM de ce complexe, notamment dans le cadre de la sclérose latérale amyotrophique. Ce projet est la suite logique de nos études sur la protéine C9ORF72 (Sellier et al., EMBO Journal 2016 ; Boivin et al., EMBO Journal 2020).
- Enfin, nous nous intéressons depuis peu à mieux comprendre les mécanismes moléculaires et cellulaires de dégradation des mitochondries altérées par autophagie (un mécanisme connu sous le nom de mitophagie) et ses dérégulations dans des maladies neurodégénératives.
Collaborations et réseaux
- Cecile MARTINAT (I-Stem, Paris), iPS and motor neurons cells models.
- Edor KABASHI (Imagine, Paris), Zebrafish animal models.
- Denis FURLING (Institut de Myologie, Paris), Pathogenic mechanisms in myotonic dystrophies.
- Patrick SCHULTZ (IGBMC, Strasbourg), CryoEM structure of the C9ORF72 protein complex.
Financements et partenaires

- 2026 - Fondation de France.
How Polyglycine Aggregates Cause Neurodegenerative Diseases - 2025 - ANR. French National Research Agency.
Investigating the role of ARLs and C9ORF72 during Immune Dysregulation - 2025 - FRM. Foundation for Medical Research.
An integrated approach to understand the pathogenic mechanisms in ALS-FTD. - 2025 - FRM pre-maturation. Foundation for Medical Research.
Therapeutical proof of concept to inhibit expression of a toxic protein in OPDM/NIID. - 2024 - AFM. French Foundation Against Myopathies.
Decipher the mechanisms underlying muscle weakness in Myotonic Dystrophy. - 2023 - ANR. French National Research Agency.
Structure & function of the C9ORF72 protein - 2021 - Foundation for Medical Research (FRM).
Identification of a novel class of human genetic disease: the polyglycine (polyG) diseases. - 2020 – Association pour la Recherche contre la Sclérose Latérale Amyotrophique (ARSLA).
Molecular mechanism and therapeutic approach for a 2-hits model of toxicity in Amyotrophic Lateral Sclerosis (ALS). - 2018 - French National Research Agency (ANR).
Identification of novel mutations in Amyotrophic Lateral Sclerosis (ALS). - 2017 - Thierry Latran Charity Foundation (TLF).
Therapeutic approaches for C9ORF72-Amyotrophic Lateral Sclerosis (ALS). - 2016 - French National Research Agency (ANR).
Role of C9ORF72 in Amyotrophic Lateral Sclerosis (ALS). - 2014 - French National Research Agency (ANR).
Study of microRNA and mitochondrial alterations in Fragile X Tremor Ataxia Syndrome (FXTAS). - 2012 - European Research Council (ERC) Starting grant.
Understanding the RNA gain of function diseases. - 2010 - French Foundation Against Myopathies (AFM).
Mis-regulation of non-coding RNA in myotonic dystrophies. - 2010 - European ERA-Net for Research on Rare Diseases (E-Rare).
Understanding the causes of heart defects in myotonic dystrophies. - 2008 - French Foundation Against Myopathies (AFM).
Understanding the causes of muscular weakness in myotonic dystrophies. - 2006 - INSERM AVENIR.
Understanding the molecular causes of myotonic dystrophies.
Actualités

NICOLAS CHARLET-BERGUERAND : PRIX JEAN VALADE, FONDATION DE FRANCE
Le 19 mars dernier, le professeur Nicolas Charlet-Berguerand a reçu le Prix Chercheur senior de la Fondation de France / Jean Valade.
Lire la suite
Prix/Distinctions

- 2026 - Prix Jean Valade de la Recherche Médicale de la Fondation de France
- 2024 - Award of scientific excellence. INSERM prime d'excellence scientifique.
- 2018 - Award of scientific excellence. INSERM prime d'excellence scientifique.
- 2012 - European Research Council Starting grant.
- 2012 - INSERM DR2. Group leader permanent position.
- 2011 - Award of scientific excellence. INSERM prime d'excellence scientifique.
- 2006 - INSERM CR1. Scientist permanent position.
- 2006 - Award Georges Deflandre. Foundation de France.
- 2006 - INSERM AVENIR. Group starting position & funding.
Publications
2026
Article dans une revue
‘Non-coding’ GGC repeats are translated into toxic polyglycine proteins in neuromuscular diseases
- Nicolas Charlet-Berguerand
- Manon Boivin
Nature Genetics ; Volume: 58 ; Page: 475-476
Article dans une revue
GGC repeat expansions within new open reading frames are translated into toxic polyglycine proteins in oculopharyngodistal myopathy
- Manon Boivin
- Jiaxi Yu
- Nobuyuki Eura
- Léa Schmitt
- David Pietri
- Erwan Grandgirard
- Patrice Goetz-Reiner
- Damien Plassard
- Chadia Nahy
- Anne Maglott
- Bastien Morlet
- Chao Gao
- Elise Lefebvre
- Muriel Philipps
- Pascal Eberling
- Angélique Pichot
- Paola Rossolillo
- Christelle Thibault
- Mustapha Oulad-Abdelghani
- Ichizo Nishino
- ...
Nature Genetics ; Volume: 58 ; Page: 517-529
2024
Article dans une revue
An unexpected polyglycine route to spinocerebellar ataxia
- Nicolas Charlet-Berguerand
Nature Genetics ; Volume: 56 ; Page: 1039-1041
2023
Article dans une revue
A biallelic loss of function variant in HORMAD1 within a large consanguineous Turkish family is associated with spermatogenic arrest
- Ozlem Okutman
- Manon Boivin
- Jean Muller
- Nicolas Charlet-Berguerand
- Stephane Viville
Human Reproduction ; Volume: 38 ; Page: 306-314
2022
Article dans une revue
PolyGA targets the ER stress-adaptive response by impairing GRP75 function at the MAM in C9ORF72-ALS/FTD
- Federica Pilotto
- Alexander Schmitz
- Niran Maharjan
- Rim Diab
- Adolfo Odriozola
- Priyanka Tripathi
- Alfred Yamoah
- Olivier Scheidegger
- Angelina Oestmann
- Cassandra Dennys
- Shrestha Sinha Ray
- Rochelle Rodrigo
- Stephen Kolb
- Eleonora Aronica
- Stefano Di Santo
- Hans Rudolf Widmer
- Nicolas Charlet-Berguerand
- Bhuvaneish Selvaraj
- Siddharthan Chandran
- Kathrin Meyer
- ...
Acta Neuropathologica ; Volume: 144 ; Page: 939-966
Article dans une revue
CGG repeat expansion in NOTCH2NLC causes mitochondrial dysfunction and progressive neurodegeneration in Drosophila model
- Jiaxi Yu
- Tongling Liufu
- Yilei Zheng
- Jin Xu
- Lingchao Meng
- Wei Zhang
- Yun Yuan
- Daojun Hong
- Nicolas Charlet-Berguerand
- Zhaoxia Wang
- Jianwen Deng
Proceedings of the National Academy of Sciences of the United States of America ; Volume: 119
Article dans une revue
De novo truncating NOVA2 variants affect alternative splicing and lead to heterogeneous neurodevelopmental phenotypes
- Marcello Scala
- Nathalie Drouot
- Suzanna C. Maclennan
- Marja W. Wessels
- Magdalena Krygier
- Lisa Pavinato
- Aida Telegrafi
- Stella A. de Man
- Marjon van Slegtenhorst
- Michele Iacomino
- Francesca Madia
- Paolo Scudieri
- Paolo Uva
- Thea Giacomini
- Giulia Nobile
- Maria Margherita Mancardi
- Ganna Balagura
- Giovanni Battista Galloni
- Alberto Verrotti
- Muhammad Umair
- ...
Human Mutation ; Volume: 43 ; Page: 1299-1313
Article dans une revue
Trinucleotide CGG Repeat Diseases: An Expanding Field of Polyglycine Proteins?
- Manon Boivin
- Nicolas Charlet-Berguerand
Frontiers in Genetics ; Volume: 13 ; Page: 843014
Article dans une revue
Evidence for a fragile X messenger ribonucleoprotein 1 ( FMR1 ) mRNA gain‐of‐function toxicity mechanism contributing to the pathogenesis of fragile X‐associated premature ovarian insufficiency
- Roseanne Rosario
- Hazel L. Stewart
- Nila Roy Choudhury
- Gracjan Michlewski
- Nicolas Charlet-Berguerand
- Richard A. Anderson
FASEB Journal ; Volume: 36 ; Page: e22612
2021
Article dans une revue
Translation of GGC repeat expansions into a toxic polyglycine protein in NIID defines a novel class of human genetic disorders: The polyG diseases
- Manon Boivin
- Jianwen Deng
- Véronique Pfister
- Erwan Grandgirard
- Mustapha Oulad-Abdelghani
- Bastien Morlet
- Frank Ruffenach
- Luc Negroni
- Pascale Koebel
- Hugues Jacobs
- Fabrice Riet
- Anke Dijkstra
- Kathryn Mcfadden
- Wiley Clayton
- Daojun Hong
- Hiroaki Miyahara
- Yasushi Iwasaki
- Jun Sone
- Zhaoxia Wang
- Nicolas Charlet-Berguerand
Neuron ; Volume: 109 ; Page: 1825-1835.e5
Page 1 sur 6