
Transcription activée
Responsables de sous-groupe : Adam BEN SHEM , Gabor PAPAI
Équipe(s) : Co-activateurs transcriptionnels
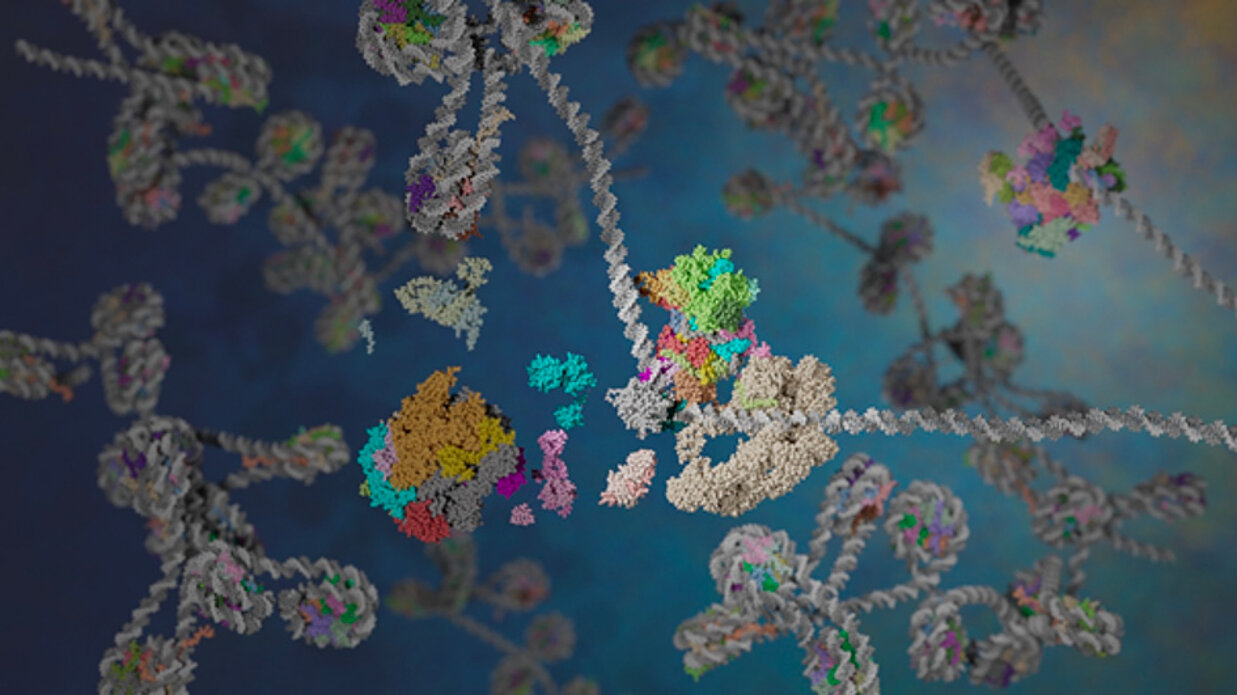
Notre groupe cherche à comprendre les mécanismes d'expression des gènes au niveau transcriptionnel et à déchiffrer les différents degrés d'organisation qui déterminent le réglage fin de la transcription lors de l'initiation.
Chez les eucaryotes, la transcription des gènes codant pour des protéines est initiée par le dépôt contrôlé de la protéine de liaison à la boite TATA, TBP, sur les promoteurs des gènes, suivi par l'assemblage ordonné d'un complexe de pré-initiation composé de sept facteurs généraux de transcription (GTF) comprenant l'ARN polymérase II (Pol II). In vivo, ce processus se déroule dans un contexte chromatinien dans lequel l'ADN est organisé en nucléosomes ? La chromatine entretient un état répressif général où les séquences régulatrices sont partiellement occluses. Par ailleurs, des protéines activatrices de la transcription se lient de manière séquence-dépendante en amont du promoteur principal afin d'adapter les niveaux de synthèse d'ARN messager aux signaux environnementaux. Les protéines activatrices recrutent des complexes coactivateurs multifonctionnels qui modifient la structure de la chromatine, agissent comme des facteurs de pontage ou de stabilisation avec les GTF et aident à déposer les TBP sur les promoteurs des gènes. L'action des co-activateurs n'est pas seulement requise pour initier la transcription, mais ces complexes jouent également un rôle majeur dans les différentes voies de réparation de l'ADN.
Nous étudions la structure et la fonction des complexes coactivateurs transcriptionnels de levure TFIID et SAGA, qui lient TBP et le déposent sur les promoteurs de gènes de manière parfaitement contrôlée. Ces deux complexes contiennent un mécanisme similaire de liaison à TBP composé d'un octamère déformé de protéines contenant des repliements d'histones. La deuxième classe de co-activateurs étudiée dans notre équipe modifie la structure de la chromatine en acétylant les queues d'histones et d'autres protéines nucléaires. SAGA et NuA4 sont des exemples prototypiques de coactivateurs contenant une activité d'acétyltransférase d'histone couplée à d'autres fonctions, spécifiques à chaque complexe. Plus récemment, notre équipe s'est penchée sur l'organisation structurelle d'une troisième classe de coactivateurs qui remodèle les nucléosomes de manière ATP-dépendante. Nous étudions la famille des remodeleurs de chromatine de la famille SWI/SNF afin de comprendre leur mode d'action et la façon dont ils sont recrutés sur leur cible génomique. La fonction de ces assemblages moléculaires est couplée à des changements de conformation et à des mécanismes de régulation allostérique. Nous cherchons à analyser le comportement dynamique des co-activateurs en combinant la cryo-microscopie électronique à haute résolution et des expériences de FRET.
La transcription et la réparation de l'ADN sont deux processus biologiques fondamentaux qui ont un fort impact sur la santé humaine, en particulier pour notre compréhension de l'origine et de la progression des tumeurs. Apparemment distincts, ces deux processus sont en fait interconnectés et partagent les mêmes mécanismes moléculaires. Le facteur de transcription/réparation TFIIH a longtemps été identifié comme un acteur commun faisant le pont entre l'étape d'initiation de la transcription et la voie de réparation par excision de nucléotides (NER) en fournissant les hélicases nécessaires pour dérouler l'ADN autour des lésions afin de les faire réparer. Plus récemment, cette interconnexion entre les mécanismes de transcription et de réparation est apparue encore plus complexe puisqu'il a été constaté que tous les facteurs de réparation étaient recrutés lors d'importants changements transcriptionnels dans la différenciation ou la reprogrammation cellulaire. Bien que des études structurales et biophysiques récentes aient conduit à un progrès considérable concernant l'architecture de TFIIH, notre compréhension de ses différents états fonctionnels et de ses interactions avec ses partenaires dans la transcription et la réparation de l'ADN est encore limitée.
Chez les invertébrés, les coactivateurs jouent des rôles importants dans le développement, la différenciation cellulaire, le maintien des cellules STEM, l'apoptose ou le vieillissement, et sont impliqués dans plusieurs maladies génétiques et le cancer. Pour élucider les mécanismes d'expression des gènes et leur dérégulation dans les pathologies humaines, nous développons des stratégies de purification pour étudier les complexes humains. Les structures à haute résolution des co-activateurs humains sont nécessaires pour éclairer leurs mécanismes moléculaires, aider à comprendre le rôle des mutations retrouvées dans les maladies et fournir un cadre pour évaluer l'action des inhibiteurs des activités enzymatiques ou des interactions protéine-protéine.
Nous utilisons une approche de biologie structurale multi-échelles pour étudier les co-activateurs qui sont pleinement actifs dans un environnement chromatinien. Nous reconstituons in vitro des systèmes nucléoprotéiques assemblés sur des matrices chromatiniennes et déterminons leur structure par cryo-microscopie électronique de particules uniques. Nous cherchons également à analyser la localisation, la structure et l'environnement moléculaire des facteurs de transcription dans le contexte du noyau cellulaire pleinement fonctionnel. Nous analysons le comportement dynamique de ces complexes dans les cellules vivantes par des approches de microscopie à fluorescence. Nous sondons l'environnement nucléaire de ces complexes par tomographie cryo-électronique de cellules cryo-immobilisées et en utilisant des sondes d'or couplées à des anticorps pour localiser les protéines d'intérêt.
Pour répondre à ces questions difficiles, nous mettons en œuvre et profitons des développements technologiques les plus avancés et participons activement aux nouvelles avancées méthodologiques dans la préparation des échantillons (systèmes d'expression dans les cellules de mammifères, méthodes de cryo-coupe, protocoles de congélation à haute pression), la microscopie cryo-électronique (nouveaux détecteurs, platines froides ultra-stables, collecte automatisée des données, correcteurs Cs, images filtrées en énergie, plaques de phase, imagerie FIB/SEM), l'analyse des images (grilles informatiques parallélisées, nouveaux algorithmes d'alignement ou de segmentation en 3-D, tri d'ensembles de données hétérogènes) et l'intégration des données (microscopie optique et électronique corrélative à haute résolution, combinaison d'informations hétérologues).
Responsables de sous-groupe : Adam BEN SHEM , Gabor PAPAI
Équipe(s) : Co-activateurs transcriptionnels
Responsable de sous-groupe : Arnaud POTERSZMAN
Équipe(s) : Co-activateurs transcriptionnels
L'assemblage du complexe de préinitiation de la transcription (PIC), composé de facteurs généraux (GTF) et de l'enzyme Pol II, sur les promoteurs de gènes est une étape de régulation clé qui sélectionne le gène à exprimer. L'assemblage du PIC sur un promoteur contenant une boîte TATA a été reconstitué in vitro et implique l'interaction de la protéine de liaison TATA (TBP) avec sa séquence promotrice correspondante, la liaison ultérieure des GTF, TFIIA et TFIIB, qui recrutent Pol II avec les autres GTF : TFIIE, TFIIF et TFIIH. Des structures atomiques du PIC reconstitué in vitro ont été déterminées récemment.
Cependant, in vivo, la TBP s'associe à 13 (14 chez la levure) facteurs associés à la TBP (TAFs) et forme le facteur de transcription TFIID qui non seulement nucléé la formation du PIC, agit également comme un co-activateur de transcription en interagissant avec les activateurs de transcription. Il est important de noter que la structure des PICs contenant TFIID n'est pas connue, ce qui laisse la possibilité que la composition et l'architecture des PICs assemblés in vivo puissent différer de ceux reconstitués in vitro.
Pour mieux comprendre le rôle du TFIID natif dans la formation des PICs endogènes et l'initiation de la transcription qui en découle, nous cherchons à :
Ces informations, combinées à l'occupation des TAF, TBP, GTF et Pol II à l'échelle du génome et aux ensembles de données sur le profilage de l'expression génétique, permettront d'identifier des ensembles distincts d'assemblages de PIC contenant des TFIID sur différentes classes de promoteurs qui sont impliqués dans l'initiation de la transcription.
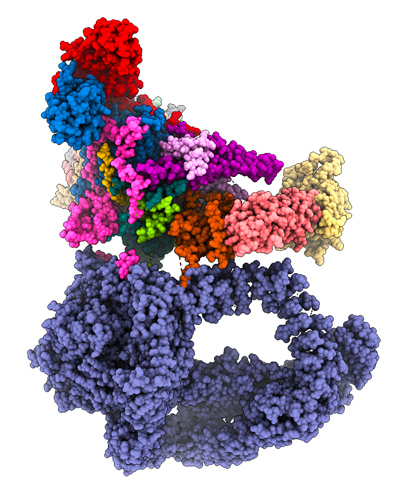
Notre objectif est de comprendre le rôle fonctionnel, l'organisation structurale et le mode d'action du coactivateur SAGA; un complexe protéique contenant 19 sous-unités et totalisant un poids moléculaire de 1,9 MDa. SAGA contient deux activités enzymatiques capables d’acétyler ou de deubiquitiner les extrémités N-terminales des histones par respectivement les sous-unités GCN5 et USP22. L’inactivation de ces activités enzymatiques conduit à une létalité embryonnaire et des mutations responsables de neurodégénérescences congénitales ou contribuant à la tumorigénèse ont été décrites dans les gènes humains codant pour ces sous-unités. Un module dédié de SAGA est responsable de son interaction avec la protéine de liaison à la TATA-box (TBP), un élément clé du complexe de pré-initiation de transcription. La grande sous-unité TRA1, d’un poids moléculaire de 400 kDa, est impliquée dans l’interaction avec les activateurs transcriptionnels et fait ainsi le lien avec les voies de signalisation cellulaire.
Notre équipe de recherche a joué un rôle clé dans la caractérisation structurale du complexe SAGA humain et de levure. Cependant, l'impact des multiples activités de reconnaissance et enzymatiques associées à SAGA sur l'expression des gènes reste mal compris. Nous cherchons à comprendre les effets respectifs de la reconnaissance de la chromatine, des activités catalytiques, de la liaison aux activateurs et à TBP, sur l'efficacité de la transcription. Le mécanisme par lequel la liaison d’un activateur à TRA1 déclenche l’assemblage du PIC et augmente la production d'ARNm est actuellement inconnu. L’interaction avec les activateurs implique des changements conformationnels coordonnés, et non encore caractérisés, dans la structure de SAGA. Il est important de comprendre comment SAGA peut réguler à la fois positivement et négativement l’initiation de la transcription d’un même gène. Un exemple ce double rôle de SAGA est illustré dans la levure S. pombe, où les cellules arrêtent de proliférer en réponse à la pénurie d’éléments nutritifs et se différencient en spores. Dans des conditions riches, SAGA réprime ces promoteurs, alors qu’en cas de privation, SAGA active la transcription des mêmes gènes. Nous cherchons à découvrir le mécanisme moléculaire par lequel SAGA intègre les signaux environnementaux pour réguler l'expression de gènes soit positivement ou négativement.
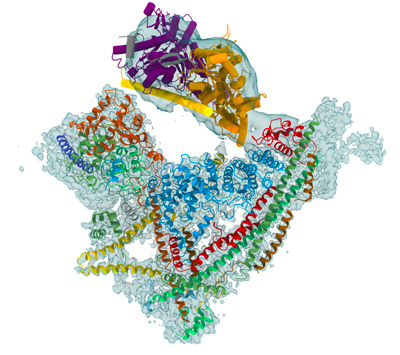
L'architecture de la chromatine est un niveau d'organisation clé pour réguler l'expression du génome, la réplication et la maintenance de l'ADN, ainsi que le contrôle épigénétique. Les remodeleurs de la chromatine utilisent l'énergie de l'hydrolyse de l'ATP pour agir sur l'unité fondamentale de la chromatine, le nucléosome, et déstabiliser l'interaction de l'octamère d'histone avec l'ADN. L'action des remodeleurs permet aux nucléosomes d'être mis en phase par rapport à leurs voisins, d'être repositionnés, échangés ou expulsés de séquences d'ADN régulatrices clés. Les remodeleurs régulent l'accès de machines moléculaires spécifiques aux séquences d'ADN génomique et modulent ainsi les principaux processus nucléaires, notamment la transcription, la réplication et la réparation de l'ADN.
Le complexe de remodelage SWI/SNF de la levure a un poids moléculaire d'environ 1,1 MDa. Il comprend la sous-unité catalytique Snf2, une hélicase qui couple l'énergie dérivée de l'hydrolyse de l'ATP au remodelage de la chromatine, ainsi que 10 sous-unités supplémentaires. Si la fonction de l'hélicase Snf2 est bien documentée, le rôle des sous-unités supplémentaires est mal connu. Malgré l'importance biologique clé des remodelages de la chromatine, il nous manque un cadre structural pour comprendre le mécanisme ATP-dépendant par lequel le complexe SWI/SNF mobilise ou perturbe le nucléosome. Plusieurs modèles ont été proposés pour le mécanisme de glissement du nucléosome et la réaction d'éviction est toujours en débat.
L'éviction des nucléosomes du promoteur pour former une région appauvrie en nucléosomes qui favorise l'initiation de la transcription est un mécanisme généralement accepté pour expliquer l'effet positif de SWI/SNF sur l'expression des gènes. Les mécanismes par lesquels SWI/SNF est dirigé vers ses sites fonctionnels dans le génome restent mal documentés. Il a été démontré que les sous-unités Swi2, Swi5 et Swi1 interagissent avec des activateurs de transcription et peuvent recruter le remodeleur vers les promoteurs de gènes en réponse aux voies de signalisation cellulaire. Le remodeleur peut également être recruté en tirant parti des marques épigénétiques sur les queues d'histones. En particulier, le bromodomaine de Snf2 est responsable de l'ancrage indépendant de l'activateur de SWI/SNF aux nucléosomes acétylés du promoteur. Les détails moléculaires de ces interactions sont mal décrits et le couplage physique entre les activités de recrutement et de remodelage n'est pas encore compris.
Un noyau de sous-unités SWI/SNF est conservé au cours de l'évolution, mais le complexe s'est diversifié de la levure aux mammifères pour atteindre une masse moléculaire de 2 MDa dans les complexes humains PBAF et BAF. La composition en sous-unités des complexes humains est spécifique du type cellulaire en raison de l'expression différentielle de gènes paralogues pour plusieurs sous-unités. Il est généralement admis que différentes combinaisons de sous-unités confèrent une spécificité cellulaire et permettent à SWI/SNF d'interagir avec des facteurs de transcription spécifiques des tissus pour réguler l'expression de gènes cibles. Des données structurales à haute résolution des complexes BAF humains endogènes constitueraient une avancée majeure dans la compréhension du mécanisme d'action des complexes humains, et de leur rôle dans le ciblage de la chromatine.
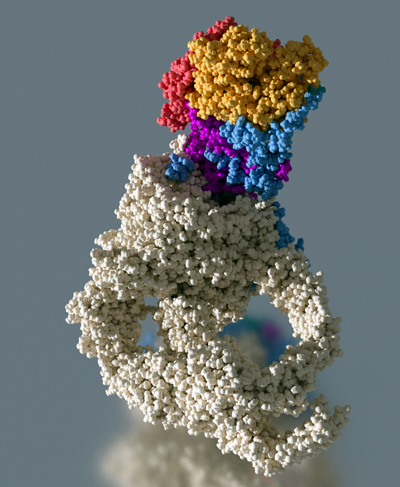
Nous souhaitons mieux comprendre le rôle fonctionnel, l'organisation structurelle et le mode d'action du co-activateur transcriptionnel NuA4, un complexe multi-protéique formé de 13 sous-unités distinctes avec un poids moléculaire total de 1,0 MDa. NuA4 intègre la signalisation épigénétique en lisant et en écrivant les modifications post-traductionnelles des histones, interagit avec les activateurs transcriptionnels et participe à l’incorporation de variants d'histones. La sous-unité Esa1 de NuA4 est une acétyltransférase essentielle à la croissance de la levure et capable de modifier l'histone H4. La grande sous-unité Tra1 de 430 kDa est impliquée dans la liaison des activateurs de transcription, faisant ainsi le lien avec les voies de signalisation cellulaire. NuA4 participe également au recrutement du variant d'histone H2A.Z, un marqueur des promoteurs de gènes actifs. Pour réaliser cette fonction, NuA4 interagit avec le remodeleur de chromatine SWR1. Chez l'homme, les homologues NuA4 et SWR1 sont physiquement associés pour former le complexe TIP60/p400, qui englobe à la fois les activités d'histone acétyltransférase (TIP60) et d'échange d'histones (p400).
L'organisation structurale des complexes complets NuA4 et SWR1 est mal comprise et nous visons à résoudre leur structure à haute résolution par cryo-EM. Le mécanisme par lequel ces facteurs sont recrutés aux promoteurs de gènes est actuellement inconnu et nous étudions les complexes supramoléculaires entre NuA4 et, à la fois, les activateurs transcriptionnels et les nucléosomes pour comprendre le réseau d'interactions qui cible NuA4 vers les gènes actifs. L'activité d'échange de l'histone H2A.Z n'a pas été étudiée jusqu'à présent au niveau structural. Nous étudions la structure des complexes NuA4 et SWR1 par cryo-EM ainsi que les intermédiaires structuraux de la réaction d'échange. Nous analysons la structure et la fonction des complexes homologues humains TIP60/p400 afin de mieux comprendre le mécanisme d'échange de H2A.Z.
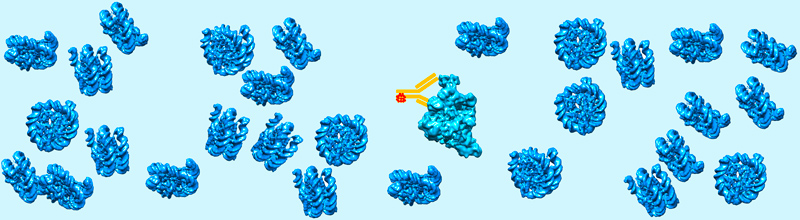
Les fonctions des macromolécules biologiques sont généralement étudiées après leur purification et leur séparation des autres composants cellulaires. Cette approche est réductionniste dans la mesure où des propriétés biologiques importantes telles que la compartimentation sans membrane, la séparation de phases, la formation de condensats, les interactions faibles ou transitoires ne se produisent que dans l'environnement encombré de la cellule intacte. Ces concepts, familiers aux sciences des polymères, conduisent ou régulent des processus cellulaires importants tels que l'expression des gènes ou la division cellulaire en créant des micro-environnements dynamiques avec des propriétés fonctionnelles dédiées. Ces phénomènes ont été décrits par imagerie des cellules vivantes mais doivent être caractérisés au niveau moléculaire. Des approches de cryo-EM ont été développées pour visualiser les assemblages moléculaires dans leur environnement cellulaire naturel. Des conditions optimales de préparation d'échantillons proches des conditions physiologiques peuvent être obtenues à l'aide de techniques de cryo-immobilisation et une reconstruction tridimensionnelle (3-D) de la cellule peut être réalisée par cryo-Tomographie Electronique (cryo-ET) de fines tranches cellulaires produites par cryo-section ou par usinage de lamelles par un faisceau ionique focalisé (FIB). La possibilité de visualiser des complexes moléculaires dans leur environnement cellulaire natif ouvre de nouvelles voies de recherche pour comprendre leur fonction cellulaire.
Un défi actuel consiste à identifier les assemblages macromoléculaires de faible abondance cellulaire ou trop petits pour être reconnus par leur forme. Pour relever ce défi, nous avons synthétisé en collaboration avec le Dr Guy Zuber (UMR7242), de nouvelles sondes à base d'or pour identifier spécifiquement les molécules d'intérêt. Les particules d'or sont couplées à des dérivés d'anticorps, délivrés dans des cellules vivantes afin de marquer sélectivement les protéines cibles. Nous cherchons à visualiser les particules d'or denses aux électrons dans des sections de cellules par cryo-tomographie électronique.

Marqueurs fonctionnalisés à base d’or pour visualiser des biomolécules et des ligands par microscopie électronique
Un projet en collaboration visant à développer de nouvelles sondes denses aux électrons pour détecter et identifier les biomolécules dans leur environnement cellulaire.
En collaboration avec Dr Guy ZUBER (UMR7242, ESBS, Illkirch, france) et Dr Ovidiu ERSEN (UMR7504, IPCMS, Strasbourg, France).

Ce réseau multidisciplinaire national vise à fournir une caractérisation multi-échelle de la cascade oncogénique, du niveau atomique à l'organisation dynamique de la cellule en réponse à des mutations génétiques, des changements environnementaux ou des modifications épigénétiques. fr.nanotumor.fr
Programme fédérateur Aviesan (PFA) - Vers un atlas cellulaire tumoral
Un programme de recherche coordonné par Jacky G. Goetz (U1109 - Strasbourg) et Patrick Schultz (IGBMC - Illkirch), et composé de 13 équipes travaillant selon quatre axes :
Axe 1 : Description moléculaire et structurale des complexes multiprotéiques initiant le processus oncogénique.
Axe 2 : Établissement d'un atlas morphologique subcellulaire au cours de la carcinogenèse : Des organelles aux réseaux moléculaires qui pilotent le cancer
Axe 3 : Biocapteurs, inhibiteurs protéiques et chimiques
Axe 4 : Intégration in vivo de complexes protéiques candidats ciblables dans des échantillons de patients
Structure de complexes multi-protéiques impliqués dans la transcription, la régulation épigénétique et la réparation de l’ADN.
Marqueurs conjugués à des particules d’or pour visualiser des complexes protéiques dans la cellule par cryo tomographie électronique.
![]()
![]()

![]()

Scientific Reports ; Volume: 12 ; Page: 2030
iScience ; Volume: 25 ; Page: 105357
iScience ; Volume: 25 ; Page: 105357
Insoluble Proteins ; Volume: 2406 ; Page: 281-317
Scientific Reports ; Volume: 12 ; Page: 2030
Multiprotein Complexes ; Volume: 2247 ; Page: 39-57
Multiprotein complexes ; Page: 17-38
Nanoscale Advances ; Volume: 3 ; Page: 6940-6948
Journal of Molecular Biology ; Volume: 433 ; Page: 166964
FEBS Journal ; Volume: 288 ; Page: 3135-3147
Page 2 sur 13