RESPONSABLE DE SOUS-GROUPE

Yann HERAULT
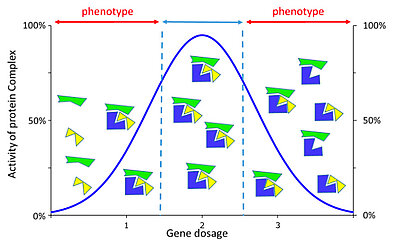 Le déséquilibre de dose peut pertuber la formation d’un complexe protéique en cas de sous- ou de sur dosage (d’après Duchon et Hérault 2016).
Le déséquilibre de dose peut pertuber la formation d’un complexe protéique en cas de sous- ou de sur dosage (d’après Duchon et Hérault 2016).
Les variantes du nombre de copies de gènes ont un rôle important dans l'apparition des troubles neurodéveloppementaux. En particulier, les syndromes 16p11.2 correspondent à une délétion ou une duplication de 600 kb de BP4-BP5 et ont été associés à un retard de développement et troubles du spectre autistique. Nous avons conçu des modèles murins portant une délétion (Del/+) ou une duplication (Dup/+) de la région Sult1a1-Spn homologue au locus humain 16p11.2 BP4-BP5. Alors que la conséquence comportementale du dosage génétique de la région 16p11 était similaire chez la souris et l'homme avec des altérations de l'activité et de la mémoire induites par la délétion, les défauts métaboliques étaient opposés (Arbogast et al., 2016). Nous avons étudié les changements craniofaciaux et constaté que les effets miroirs étaient répliqués et conservés entre les modèles humains, souris et rats (Qiu et al., 2019) avec de nombreux gènes impliqués. D'autres études ont mis en évidence l'implication du déséquilibre génétique Kctd13 dans la délétion 16p11.2 via la régulation de la voie RHOA. En parallèle, nous avons généré un nouveau modèle de souris avec une petite suppression de deux exons clés dans Kctd13. Ensuite, nous avons ciblé la voie RHOA pour sauver les phénotypes cognitifs des modèles murins de délétion Kctd13 et 16p11.2 dans un contexte génétique pur. Nous avons utilisé une administration chronique de fasudil (HA1077), un inhibiteur de la Rho-associated protein kinase (ROCK), pendant six semaines, dans des modèles murins portant une inactivation hétérozygote de Kctd13, ou la délétion de l'ensemble 16p11.2 BP4-BP5 région homologue. Nous avons constaté que la petite délétion hétérozygote Kctd13 induit un phénotype cognitif similaire à l'ensemble de la délétion de la région homologue 16p11.2, chez les souris Del/+. Ensuite, nous avons montré que le traitement chronique au fasudil peut restaurer la mémoire de reconnaissance d'objets chez des souris mutantes hétérozygotes adultes pour la délétion Kctd13 et 16p11.2. De plus, l'amélioration de l'apprentissage et de la mémoire était parallèle au changement du parcours RHOA (Martin Lorenzo et al., 2021).
Le syndrome de Koolen de Vries (KdVS) est un trouble multisystémique causé soit par une microdélétion dans la région chromosomique 17q21.31, soit par une variante tronquante du gène de l'unité 1 du complexe régulateur KAT NSL (KANSL1) situé dans l'intervalle. Nous avons généré trois modèles murins pour la délétion (Del/+), la duplication réciproque (Dup/+) de la région synténique 17q21.31 et la perte de fonction hétérozygote Kansl1 pour étudier la physiopathologie. Les études de caractérisation ont confirmé le rôle de KANSL1 dans les phénotypes associés au KdVS et ont montré une transmission synaptique défectueuse et une neurogenèse altérée accompagnées de défauts dans la facilitation des impulsions appariées et d'une diminution de la ramification des cellules positives à la doublecortine dans les hippocampes adultes. Le profilage de l'expression génique a également montré que les gènes contrôlant la transmission synaptique et la neurogenèse ou mutés dans les TSA (Grid1, Cntnap2, Nrxn1, Nrxn3, Adcyap1 et Ucn) étaient mal exprimés dans les hippocampes de souris KdVS (Arbogast et al., 2017).
Le gène lié à l'X, PTCHD1, a été trouvé supprimé, ou avec des mutations tronquantes et faux-sens, chez des hommes atteints de DI et de TSA. Nous avons généré un knock-out complet de Ptchd1 chez la souris et nous avons découvert que la perte complète de fonction de Ptchd1 induisait un phénotype robuste, ainsi que des modifications du transcriptome et de la réponse électrophysiologique dans l'hippocampe, une région centrale du cerveau (Ung et al., 2018). Des changements ont été trouvés dans l'activité glutamatergique excitatrice neuronale qui provoquent un déséquilibre synaptique excitateur/inhibiteur, une hypothèse importante pour l'étiologie du TSA.
Haploinsuffisance DYRK1a et maladie du retard mental 7 : Le gène à double spécificité de la kinase 1A régulée par la phosphorylation de la tyrosine (DYRK1A) est considéré comme le principal acteur du déficit cognitif dans le syndrome de Down (DS), et son haploinsuffisance est responsable du syndrome de retard mental autosomique dominant 7 (MRD7), caractérisé par une déficience intellectuelle (DI), une microcéphalie, une épilepsie et un autisme (Duchon and Herault, 2016). Nous avons généré de nouveaux modèles de perte de fonction ou de mutation ponctuelle de Dyrk1a et également de destruction de l'activité kinase (kinase dead mutant) chez la souris et le rat qui sont actuellement à l'étude.
Autres maladies rares : Nous avons également collaboré sur d’autres maladies rares avec DI dues à un effet de dosage : syndrome de William-Beuren (Segura-Puimedon et al., 2014)) ; ou à des mutations dans un gène unique : ATP6AP2 (Dubos et al., 2015; Wendling et al., 2017), MECP-2 (Delepine et al., 2016), OPHN1 (Meziane et al., 2016), et la duplication 24 d'Arx (Dubos et al., 2018).