

Pathophysiology of Down's syndrome and rare dose-effect diseases causing intellectual disabilities (ID) or autism spectrum disorders (ASD) and other comorbidities
Team Leader : Yann HERAULT
Department : Translational medecine and neurogenetics
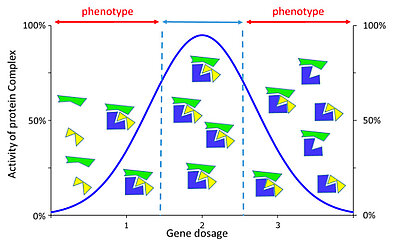 Genetic dosage imbalance can alter the formation of a protein complex (from Duchon et Hérault 2016)
Genetic dosage imbalance can alter the formation of a protein complex (from Duchon et Hérault 2016)
Gene copy number variants have an important role in the appearance of neurodevelopmental disorders. Particularly, the 16p11.2 syndromes correspond to 600 kb BP4-BP5 deletion or duplication and have been associated with developmental delay and autism spectrum disorders. We engineered mouse models carrying a deletion (Del/+) or a duplication (Dup/+) of the Sult1a1-Spn region homologous to the human 16p11.2 BP4-BP5 locus. Whereas the behavioral consequence of the 16p11 region genetic dosage was similar in mice and humans with activity and memory alterations induced in the deletion, the metabolic defects were opposite (Arbogast et al., 2016). We investigated the craniofacial changes and found that mirror effects were replicated and conserved between human, mouse and rat models (Qiu et al., 2019) with many genes implicated. A few studies highlighted the implication of Kctd13 genetic imbalance in the 16p11.2 deletion through the regulation of the RHOA pathway. In parallel, we generated a new mouse models with a small deletion of two key exons in Kctd13. Then we targeted the RHOA pathway to rescue the cognitive phenotypes of the Kctd13 and 16p11.2 deletion mouse models in a pure genetic background. We used a chronic administration of fasudil (HA1077), an inhibitor of the Rho-associated protein kinase (ROCK), for six weeks, in mouse models carrying a heterozygous inactivation of Kctd13, or the deletion of the entire 16p11.2 BP4-BP5 homologous region. We found that the small Kctd13 heterozygous deletion induced similar cognitive phenotype to the whole deletion of the 16p11.2 homologous region, in the Del/+ mice. Then we showed the chronic fasudil treatment can restore object recognition memory in adult heterozygous mutant mice for Kctd13 and for 16p11.2 deletion. In addition, the learning and memory improvement was parallel to change in the RHOA pathway (Martin Lorenzo et al., 2021).
The Koolen de Vries syndrome (KdVS) is a multi-system disorder caused either by a microdeletion in chromosomal region 17q21.31 or a truncating variant in the KAT regulatory NSL complex unit 1 (KANSL1) gene located within the interval. We generated three mouse models for the deletion (Del/+), the reciprocal duplication (Dup/+) of the 17q21.31 syntenic region and the heterozygous Kansl1 loss-of-function to investigate the pathophysiology. Characterization studies confirmed the role of KANSL1 in the KdVS-associated phenotypes and showed defective synaptic transmission and altered neurogenesis accompanied by defects in paired-pulse facilitation and decrease branching of doublecortin positive cells in the adult hippocampi. Gene expression profiling also showed that genes controlling synaptic transmission and neurogenesis or mutated in ASD (Grid1, Cntnap2, Nrxn1, Nrxn3, Adcyap1 and Ucn) were misexpressed in KdVS mouse hippocampi (Arbogast et al., 2017).
The X-linked gene, PTCHD1, has been found deleted, or with truncating and missense mutations, in male individuals with ID and ASD. We have generated a full knock-out of Ptchd1 in the mouse and we have unraveled that the complete loss-of-function of Ptchd1 induced robust phenotype, as well as changes in the transcriptome and electrophysiological response in the KO hippocampus, a central brain region for cognitive functions and social communication (Ung et al., 2018). Changes were found in neuronal excitatory glutamatergic activity that cause excitatory/inhibitory synaptic imbalance, a prominent hypothesis for the etiology of ASD.
DYRK1a haploinsufficiency and mental retardation disease 7: The dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (DYRK1A) gene is considered as one of the main drivers for the cognitive deficit in Down syndrome (DS), and its haploinsufficiency is responsible for the autosomal dominant mental retardation 7 (MRD7) syndrome, characterized by intellectual disability (ID), microcephaly, epilepsy and autism (Duchon and Herault, 2016). We have generated new models of Dyrk1a loss of function or point mutation and also killing the kinase activity (kinase dead mutant) in mouse and rat that are currently under investigation (and see project on Dyrk1a, brain development and rare diseases)
Other rare diseases: We have also focused our interest on diseases with ID due either to dosage effects: William-Beuren syndrome (Segura-Puimedon et al., 2014)); or to mutation in single genes: ATP6AP2 (Dubos et al., 2015; Wendling et al., 2017), MECP-2 (Delepine et al., 2016), OPHN1 (Meziane et al., 2016), and the duplication 24 of Arx (Dubos et al., 2018).