
Pathophysiology of neuromuscular diseases
Team Leader : Jocelyn LAPORTE
Department : Translational medecine and neurogenetics
SUBGROUP LEADER
Identify the genetic basis of neuromuscular diseases. 50% of patients are devoid of genetic diagnosis, while it is a requisite for better healthcare and genetic counselling. The identification of the implicated genes also help to better understand the pathomechanism and identify novel therapeutic targets.
Identification of the genetic basis of myopathies and other human diseases (MYOCAPTURE)
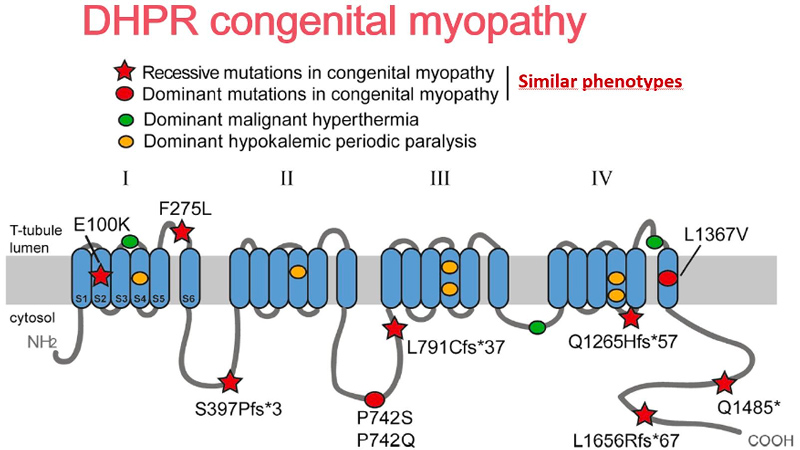
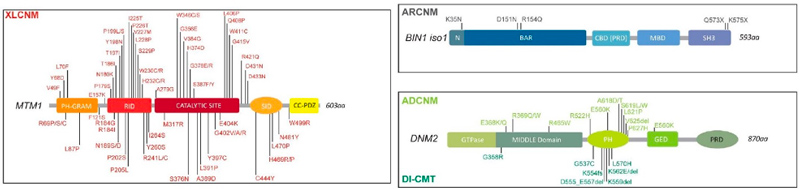
Identification of genes mutated in inherited diseases is necessary to access more specific health care and existing therapies, for familial genetic counselling and potentially further prenatal diagnosis, and for inclusion into clinical trials. Also, novel implicated genes represent novel therapeutic targets. Using positional cloning, functional candidate approach and massively parallel sequencing, we identified or participated to the identification of the first disease mutations in 20 genes: the phosphoinositides phosphatase MTM1 in myotubular myopathy (1996), the dead-phosphatase MTMR13 in peripheral neuropathy (2003), the transcriptional co-activator MAMLD1 in X-linked hypospadias (2006), the large GTPase DNM2 in dominant centronuclear myopathy (2005) and in recessive arthrogryposis (2013), the membrane remodelling protein BIN1 in recessive (2007) and dominant (2014) centronuclear myopathy and its splicing defect in myotonic dystrophy (2011), the phosphatase Jumpy/MTMR14 in centronuclear myopathy, the calcium channel RYR1 in the Samaritan myopathy (2012), the calcium sensor STIM1 in dominant tubular aggregate myopathy (2013), titin (TTN) in recessive centronuclear myopathy (2013), the unconventional myosin MYO18B in nemaline myopathy (2015), the presynaptic high-affinity choline transporter SLC5A7 in congenital myasthenic syndrome (2016), the reductase PYROXD1 in congenital myopathy (2016), the CACNA1S pore of the DHPR channel in congenital myopathy (2017), the kinase ZAK in congenital myopathy (2017), Myopalladin MYPN in nemaline myopathy (2017), the calcium buffer calsequestrin CASQ1 in tubular aggregates myopathy (2018), the actin organizer alpha-actinin 2 (ACTN2) in congenital myopathy (2019), the geranylgeranyl diphosphate (GGPP) synthase (GGPS1) in the Muscular Dystrophy/Hearing Loss/Ovarian Insufficiency Syndrome (2020), the phosphorylase PYGM in the first dominant glycogen storage disease (2020), the Myosin Chaperone UNC-45B in myopathy with eccentric cores (2020).
Examples:
Identify the genetic basis of Myalgia (GENTAUMIX)
Our objectives are to identify the genetic causes of exercise-induced myalgia (= muscle pain) with or without rhabdomyolysis by combining genomic and transcriptomic approaches in a prospective cohort of 198 well-characterized participants in whom extensive phenotype data are available. Insight into the pathomechanism will be achieved through transcriptome comparison. The possibility to work on the affected tissue (muscle) for DNA and RNA sequencing allows to obtain physiological relevant data and detect potential somatic mosaicism.
Identify the genetic basis of high athletic performances (ATHLOME)
To better understand the genetic basis of the neuromuscular and respiratory physiology, we aim to identify rare genetic variants correlating with high athletic performances. Genome sequencing and analysis are performed in homogeneous and sub-classified cohorts of athletes of national and international level. Identified genes or variants are confirmed through functional analysis in laboratory models and clinical setting. This project will allow to better understand genetic predispositions to adapt to extreme environments, and to identify novel therapeutic targets for the neuromuscular and respiratory pathologies.

Team Leader : Jocelyn LAPORTE
Department : Translational medecine and neurogenetics

Her work, supervised by Jocelyn LAPORTE and Valérie BIANCALANA is entitled :“Comparative analysis of viral and development of non-viral muscle…
Read more
Article in a journal
Acta Neuropathologica Communications ; Volume: 12 ; Page: 191
Article in a journal
Genome Medicine ; Volume: 16 ; Page: 87
Article in a journal
Journal of Neurology
Article in a journal
Nature Genetics ; Volume: 56 ; Page: 395-407
Article in a journal
Acta Neuropathologica Communications ; Volume: 10 ; Page: 101
Article in a journal
Journal of Medical Genetics ; Volume: 59 ; Page: 559-567
Article in a journal
Acta Neuropathologica Communications ; Volume: 9 ; Page: 155
Article in a journal
Genes ; Volume: 12 ; Page: 1199
Article in a journal
Clinical Genetics ; Volume: 100 ; Page: 234 - 235
Article in a journal
Neurogenetics ; Volume: 22 ; Page: 33-41