
Pathophysiology of neuromuscular diseases
Team Leader : Jocelyn LAPORTE
Department : Translational medecine and neurogenetics
SUBGROUP LEADER
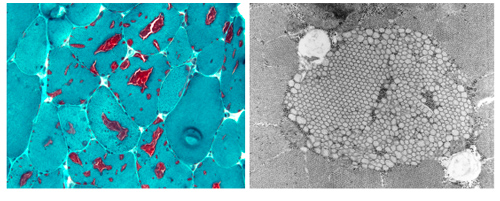
Tubular aggregate myopathy (TAM) and Stormorken syndrome (STRMK) are clinically overlapping disorders characterized by muscle weakness associated with additional multi-systemic signs including thrombocytopenia, miosis, hyposplenism, ichthyosis, short stature, and dyslexia. Muscle biopsies of TAM/STRMK patients typically show dense accumulations of membrane tubules appearing in red on Gomori staining and representing one of the main signs of ageing. Our team identified the genetic causes of TAM/STRMK, and we discovered that the mutations in the Ca2+ sensor STIM1 and the Ca2+ channel ORAI1 result in excessive extracellular Ca2+ entry. To correlate the cellular alterations with disease development, we generated and characterized a TAM/STRMK mouse model recapitulating the main clinical signs of the human disorder.
We aim to
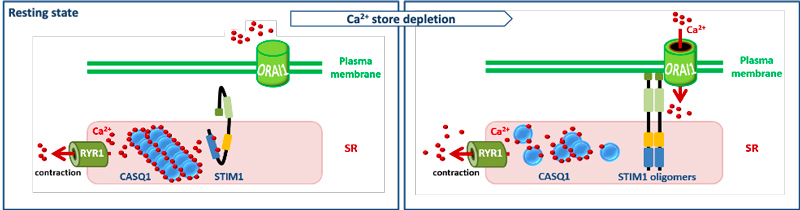
Calcium (Ca2+) is a ubiquitous second messenger implicated in the regulation of fundamental adaptive and developmental processes in all cell types, and mediates nerve conduction, hormone release, coagulation, and muscle contraction. Consistently, pathologic alterations of Ca2+ entry, Ca2+ storage, or Ca2+ release can severely impact on Ca2+ signaling and disturb various molecular, physiological, and biochemical functions in the tissues and organs, resulting in human diseases.
One of the main mechanisms regulating Ca2+ homeostasis is store-operated Ca2+ entry (SOCE). SOCE relies on the concerted activity of the reticular Ca2+ sensor STIM1 and the plasma membrane Ca2+ channel ORAI1. Our team has shown that STIM1 and ORAI1 gain-of-function (GoF) mutations induce excessive Ca2+ influx through SOCE over-activation, and cause tubular aggregate myopathy (TAM) and Stormorken syndrome (STRMK), two overlapping disorders characterized by muscle weakness and additional multi-systemic signs affecting growth, platelets, spleen, skin, and intellectual abilities (Bohm et al., 2013, Bohm et al., 2017). We also characterized a faithful mouse model for TAM/STRMK harbouring the most common STIM1 mutation, (Silva-Rojas et al., 2019), and we now plan to generate an additional ORAI1 model and investigate the pathophysiological effect of overactive SOCE on skeletal muscle, platelet, ski n, and spleen function and structure through the combination of transcriptomics with morphological and functional studies.
There is currently no therapy for TAM/STRMK. Through functional investigations in cellular and animal models, we provided the evidence that TAM/STRMK arises from aberrant Ca2+ homeostasis (Bohm et al., 2013, Bohm et al., 2017, Silva-Rojas et al., 2019). Ca2+ balance is susceptible to manipulation, and we aim to genetically modify the key factors of the SOCE pathway to control extracellular Ca2+ entry in skeletal muscle and other affected tissues. Based on this proof-of-concept, we next aim to develop pharmacological approaches to identify molecules and drugs with the potential to improve the multi-systemic TAM/STRMK phenotype.
The expected results on our cell and animal models may serve for the development of therapies for other Ca2+-related disorders affecting skeletal muscle, platelets, spleen, or skin.
Team Leader : Jocelyn LAPORTE
Department : Translational medecine and neurogenetics

Her work, supervised by Albert WEIXLBAUMER is entitled “Unravelling the early events of transcription-translation coupling in bacteria: structural…
Read more