

Integrative structural biology of SMC complexes
Subgroup Leader : Marie-Laure DIEBOLD
Teams : Molecular Basis of Chromatin and Transcription Regulation
Understand the molecular bases of epigenetic mechanisms
Understand the regulation of nuclear processes by epigenetic effectors
Understand the dysregulation of epigenetic mechanisms in diseases
Understand how to modulate the activity of epigenetic effectors by drug candidates
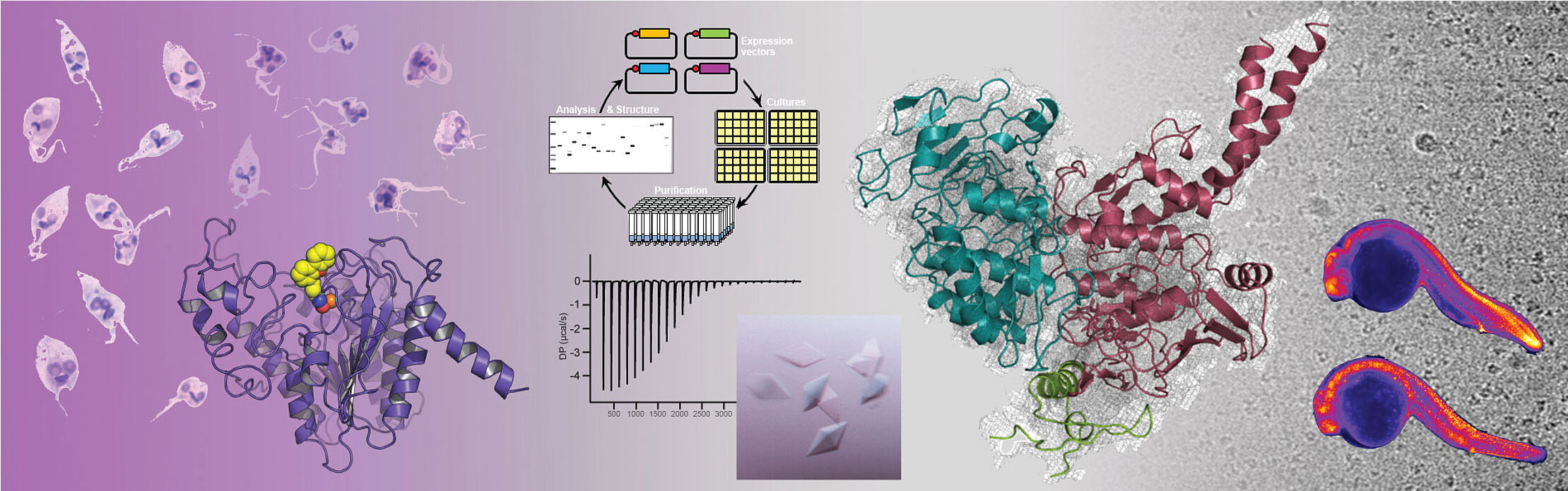
Our team studies eukaryotic epigenetic mechanisms that are essential to cell homeostasis and the development of organisms. In particular, we characterize the molecular basis of epigenetic mechanisms to decipher their involvement in the regulation of nuclear processes and understand their physiological roles. We use this knowledge to determine how mutations in epigenetic effectors can lead to many different pathologies. We are also implementing strategies to characterize small molecules that inhibit or regulate the activity of epigenetic effectors, with the aim of helping the development of drug candidates targeting epigenetic pathologies, in particular cancers, neurodevelopmental and neglected diseases (malaria, leishmaniasis, schistosomiasis, Chagas disease).
Our objectives are to combine our different lines of research to characterize epigenetic mechanisms, understand their implications in pathologies, and participate in the development of epigenetic therapies. Our studies are based on in vitro biochemical, biophysical and structural biology analyses. However, we develop all of our projects within the framework of integrative biology strategies that combine in vitro, in cellulo and in vivo analyses. In particular, we are developing within the team the use of zebrafish to address our three research axes (mechanisms, pathologies and small molecules) in an animal model to characterize the structure/function relationships of our epigenetic targets.
Our scientific projects specifically target three types of epigenetic effectors: deacetylases, histone chaperones and SMC (Structural Maintenance of Chromosomes) complexes. Our epigenetic targets are all functionally related, our ultimate goal being to characterize their physical and functional interrelationships. In support of our epigenetic scientific projects, our team has been developing for more than two decades a technology for the reconstitution of protein complexes by multi-expression in bacterial hosts. This technology and our associated know-how are essential to all of our scientific projects and are also the source of specific collaborative projects with other research teams.
PhD students
Pauline LANDWERLIN (2018-2022)
Marina VITORIA GOMES (2017-2021)
Elizabeth RAMOS MORALES (2017-2021)
Pernelle KLEIN (2016-2020)
Tajith Baba SHAIK (2014-2018)
Post-doctoral fellows
Nataliia ALEKSANDROVA (2016-2018)
Martin MAREK (2010-2017)
Régis BACK (2015-2017)
Michael KOCH (2007-2009)
Protein acetylation is a post-translational modification that regulates the activity of a very large number of cellular actors and that rivals phosphorylation in importance. In particular, acetylation plays an essential role in epigenetics by acting on chromatin compaction but also in epigenetic signaling. The acetylation mark is reversible, being deposited by lysine acetyl transferases (KATs), read by bromodomains, and removed by lysine deacetylases (KDACs). Enzymes of the acetylation cycle are important therapeutic targets, and zinc-dependent KDACs (called Histone Deacetylases, HDACs) were among the first targets of epigenetic drugs approved by the Food and Drug Administration (FDA) for the treatment of certain cancers.
Our team studies HDACs to understand their enzymatic mechanisms but also to help develop new drug candidates that can target different cancers as well as neglected diseases (malaria, leishmaniasis, schistosomiasis, Chagas disease). The active sites of each of the HDACs show many similarities but also own specificities. Thus, to date, many questions remain unanswered on the specific recognition by HDACs of their protein targets and on the selective inhibition of the different HDAC isozymes in order to develop drugs with a broader spectrum of use.
Our team initially addressed these issues by participating in two large European projects (SEtTReND and A-ParaDDisE) which brought together European, Australian and Brazilian researchers to develop drug candidates selectively targeting the HDACs of pathogenic parasites responsible for neglected diseases (malaria, leishmaniasis, schistosomiasis, Chagas disease). We were able to solve the structure in inhibited form of two pathogenic HDACs, HDAC8 of the parasitic worm Schistosoma mansoni (smHDAC8), responsible for schistosomiasis, and DAC2 of the kinetoplastid Trypanosoma cruzi (tcDAC2), responsible for Chagas disease. While the structure of smHDAC8 showed a strong resemblance to its human counterpart, certain differences allowed progress towards the development of selective inhibitors for this pathogenic enzyme.
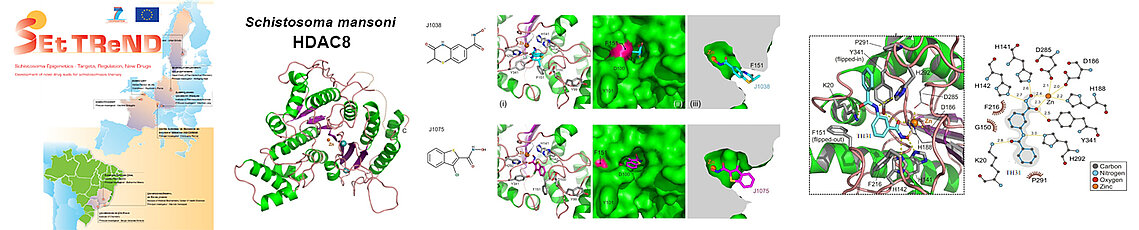
HDAC8 of the pathogenic worm Schistosoma mansoni (smHDAC8) was the major target of the European project SEtTReND (Schistosoma Epigenetics: Targets, Regulation, New Drugs). The resolution of the structure of this HDAC revealed its structural specificities. This made it possible to search for the first inhibitory molecules with a certain selectivity for smHDAC8. The structures of the first most promising inhibitors (J1038 and J1075) in complex with smHDAC8 showed their mode of binding to the enzyme. An optimization study of these compounds was then carried out to lead to a series of selective drug candidates for smHDAC8 compared to human HDACs and to reveal the structure/function relationships of this enzyme.
The structure of tcDAC2, on the other hand, revealed structural characteristics very different from human enzymes, which facilitates the development of selective drug candidates against trypanosomes, but also against the parasites responsible for malaria and leishmaniasis, these pathogens having also atypical HDACs but whose structures remain to be determined. All of our studies demonstrate that the differences in the active sites of HDACs, pathogenic but also human, form the basis for the specific recognition of their protein targets.
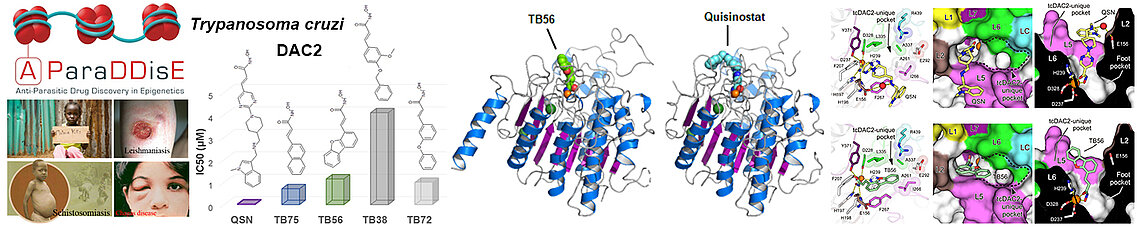
The SEtTReND project provided the proof of concept of the possibility of modifying epigenetic drugs approved by the FDA to derive selective drug candidates against eukaryotic pathogens. The A-ParaDDisE (Anti-Parasitic Drug Discovery in Epigenetics) project applied this strategy to the epigenetic enzymes of other pathogens responsible for malaria, leishmaniasis and the Chagas disease. In particular, through our integrative study of Trypanosoma cruzi DAC2 (tcDAC2), we have shown that the HDACs of these pathogens are structurally divergent from human HDACs, which will facilitate the development of drug candidates against these pathogens.
The objectives of our project on histone deacetylases is to decipher in biochemical and structural terms the specific interactions between HDACs and their target proteins, to understand the deregulation of their activity in diseases, and to modulate these interactions and these activities using small molecules of therapeutic interest.
Selected publications
The histone proteins (H2A, H2B, H3, H4) form the scaffolding on which the nucleosomes are assembled, which themselves form the basic unit of chromatin. Among the histones, the canonical histones are used in particular for the formation of chromatin during cell division, while the histone variants have specific functional roles at different times of the cell cycle and at different locations in the genome. The histones form specific pairs (H2A/H2B and H3/H4), and none of these pairs is found free, either in the cytoplasm or in the nucleus, to prevent them from being able to interact in an inopportune manner with the nucleic acids. Thus, histone pairs are complexed to histone chaperones until they are incorporated into chromatin.
Among histone chaperones, some are not specific to any particular histone pair. Some other chaperones are specific to either the H2A/H2B pair or the H3/H4 pair, regardless of their canonical or variant characteristics. Finally, other chaperones will precisely recognize a canonical pair or a specific variant pair. Histone chaperones can be involved in the transport of histone pairs between different cellular compartments or within the same compartment, while others will be involved in supply, exchange and removal of the histone pairs within chromatin. The functional importance of the various histone pairs and their correct integration within the chromatin implies understanding their interactions with their chaperones and how the latter interact with the nuclear effectors.
Our team addresses these themes by studying mainly two histone chaperones with different roles: Spt6 and the chaperones of the histone variant H2A.Z. The Spt6 chaperone travels with RNA polymerase II. Spt6 notably reconstitutes nucleosomes in the wake of this polymerase during gene transcription. Spt6 also acts as a transcription elongation factor and is coupled to the messenger RNA export system. Our studies revealed that this chaperone acts as a recruitment platform for numerous nuclear effectors during the initiation and elongation phases of transcription by RNA polymerase II. Notably, Spt6 to play its various functional roles by interacting with these nuclear effectors by interacting with them using short specific patterns.
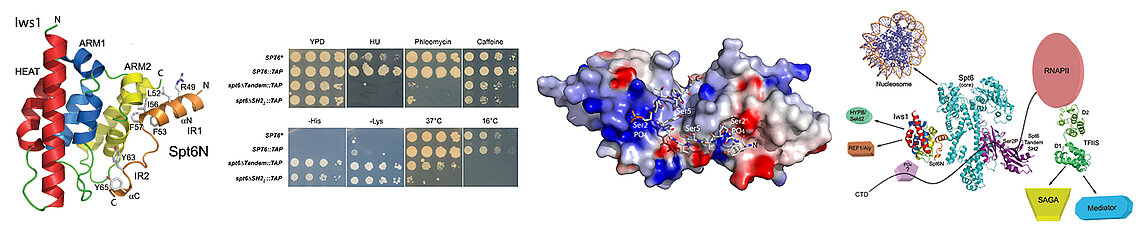
The N- and C-termini of the Spt6 protein has, at its N- and C-terminal harbour specific motifs of interaction with various nuclear effectors. We have resolved the structure of one of the N-terminal motifs of Spt6 attached to the Iws1 protein, which is involved in the maturation and export of messenger RNAs. We have also solved the structure of the C-terminal tandem of SH2 motifs of Spt6, which interacts specifically with the C-terminal tail of RNA Polymerase II and which allows Spt6 to cooperate functionally with this polymerase. We validated our structural and interaction data with yeast genetic experiments in collaboration with Fred Winston's team at Harvard. Our data demonstrate the role of interaction and signal integration platform played by Spt6.
In the case of the histone variant H2A.Z, it is recognized by two chaperones in humans, YL1 and ANP32E. In collaboration with the Hamiche team of the IGBMC, we studied the deposition and removal of the H2A.Z/H2B pair within the chromatin by these two chaperones. We have highlighted the similar mechanisms used by these two chaperones to specifically recognize the H2A.Z/H2B pair. On the other hand, our work has revealed the divergent mechanisms of these two chaperones to ensure that the H2A.Z/H2B pair is correctly deposited within the chromatin, in the case of YL1, and is removed from the chromatin by recognizing a motif buried in the heart of the nucleosome, in the case of ANP32E. This work also revealed functional links between ANP32E and the transcription factor/chromatin insulator CTCF.
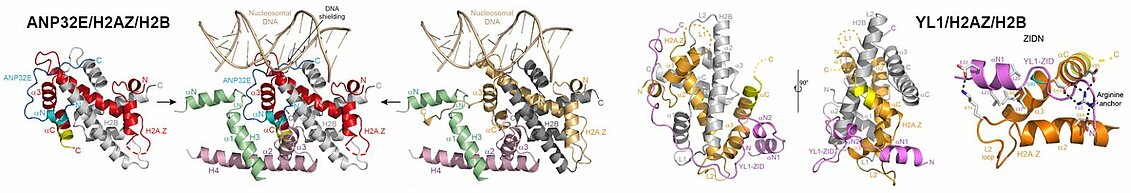
The H2AZ histone variant plays specific functional roles during the cell cycle, being found in particular in the regulatory parts of the genome when these are involved in gene transcription. The Hamiche team from the IGBMC has found two chaperones of this variant in humans: ANP32E and YL1, the first removing the H2AZ/H2B pair from chromatin, the second depositing it on the chromatin. Our structures of these chaperones linked to the H2AZ/H2B pair showed that they both recognize the H2AZ variant due to the absence of a glycine residue in its C-terminal helix and that their interaction with the H2AZ/H2B pair lengthened this C-terminal helix. If this conformational change of the C-terminal helix of H2AZ is sufficient for the eviction of H2AZ/H2B from chromatin by ANP32E, YL1 interacts more extensively and specifically with this pair, thus ensuring that it is the H2AZ/H2B pair which is correctly deposited on the chromatin.
We are pursuing this work to better understand how histone chaperones interact with other nuclear actors, to be directed specifically to certain chromatin domains and to recruit other epigenetic effectors.
Selected publications
Many nuclear processes require the spatial rapprochement of different parts of our genome as well as the formation of chromatin loops. These processes keep sister chromatids together during DNA replication, then condense them into chromosomes and distribute them evenly among daughter cells during cell division. These processes also allow DNA repair and the regulation of transcription. All of these processes are required throughout the cell cycle and depend in eukaryotes on paralogous complexes called “Structural Maintenance of Chromosomes” (SMC). In vertebrates, these include mitotic and meiotic Cohesins, Condensins I and II, and the SMC5/SMC6 complex.
The different SMC complexes have similar organizations and mechanisms. However, each has its own biological functions which are ensured with the help of specific core and regulatory subunits. To understand the functions of each of the SMC complexes and their dysregulation in diseases, it is essential to be able to distinguish the mechanisms shared by these complexes and those which are specific to them. In particular, all of the SMC complexes have an ATPase activity essential to their functions carried by the ATPase heads of each of their two core Smc subunits. How these Smc subunits bind ATP and hydrolyze it to ADP, and how this ATPase activity influences the conformation of SMC complexes, thus allowing them to perform their functional roles, remain poorly understood.
Our team studies human mitotic cohesin which is essential for sister chromatid cohesion, chromosome separation, DNA repair, regulation of transcription, and for the dynamic formation of chromatin loops and topologically associated domains (TADs). The importance of Cohesin is demonstrated by its mutation in many liquid and solid cancers and in pathologies called cohesinopathies which are neurodevelopmental diseases with autistic traits. Cohesin has specific regulatory proteins, including HDAC8 which deacetylates Cohesin during its functional cycle, but also CTCF which physically and functionally interacts with human Cohesin to form chromatin loops and TADs.
Our team studied the ATPase activity of Cohesin by combining biochemical, biophysical and structural, by crystallography and cryo-electron microscopy, in vitro analyses as well as in vivo, using zebrafish. This study allowed us to characterize at the molecular level the different stages of the ATPase cycle of human Cohesin, highlighting significant differences between vertebrate Cohesins and those of yeasts. In addition, our work has enabled us to understand the requirement for the core Cohesin complex to associate with DNA and the regulatory subunit NIPBL to have a maximum ATPase activity, the core complex having only low ATPase activity when alone or associated with DNA.
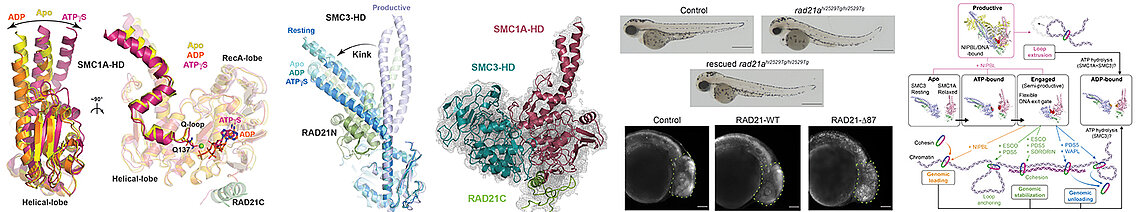
The Cohesin complex is a major player in sister chromatid cohesion, chromsome segregation and 3D genome organization . Cohesin acts through conformational changes which are induced by its ATP binding and hydrolysis activity which is modulated by different regulators. The deregulation of the activity of Cohesin by mutations of its core and regulatory subunits leads to a large number of cancers and neurodevelopmental diseases called cohesinopathies. To better understand the mode of action of human Cohesin, we characterized biochemically, biophysically and structurally the movements of its ATPase domains, carried by its core subunits SMC1A and SMC3, when these bind or hydrolyze ATP, and characterized the ATP-dependent interaction of these ATPase domains (left panels). Our different data allow us to describe the conformational changes of Cohesin during its ATPase cycle (right panel). We used zebrafish to functionally validate in vivo our biochemical and structural data (central panel).
This initial work by our team on vertebrate Cohesin opens up broad perspectives for studying the molecular mechanisms that govern the functional activity of human Cohesin, its regulations, and its involvement in cancers and cohesinopathies.
Selected publications
Our team has a long tradition of collaborating with teams from other departments within the IGBMC as well as outside of the IGBMC. Our first collaborations allowed us to address many topics around cancer. The new collaborations that we have established lead us to also study neurodevelopmental diseases. These themes are directly related to our scientific questions on Cohesin and its involvement in many cancers and in cohesinopathies.
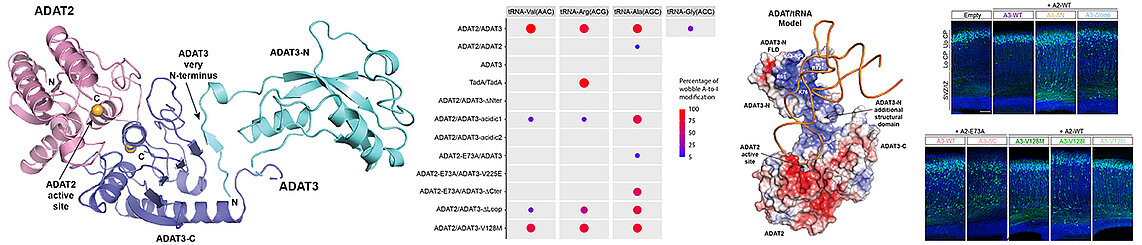
In collaboration with the teams of Juliette Godin at the IGBMC and Laurence Drouard at the IBMP, Strasbourg, we have characterized the ADAT complex which modifies the adenosine bases at the wobble position of tRNAs into inosine, which enables the extension of the genetic code genetic. In addition, the V128M mutation in the N-terminal domain of its ADAT3 subunit is responsible for intellectual disabilities and other neurodevelopmental diseases. Our structure of the murine ADAT complex and our biophysical data showed that the N-terminal domain of ADAT3 enables the specific recognition of tRNAs to present them to the catalytic domain of ADAT2. These data and our enzymatic assays showed that the N-terminal domain of ADAT3 is essential for the activity of the ADAT complex and that the V128M mutation interferes with the correct presentation of the adenosine wobble to ADAT2 without however affecting tRNA binding.
Selected publications
Many nuclear effectors, especially those involved in epigenetic mechanisms, act as multiprotein complexes that are difficult to assemble and purify. In addition, the quality and homogeneity of the samples is essential for in vitro analyses, particularly structural ones. To meet these challenges, our team has been developing for more than twenty years a technology for the co-expression in the Escherichia coli bacteria of protein complex subunits. This technology makes it possible to characterize, reconstitute and purify protein complexes, and is essential to the development of all of our projects. This technology is however extremely versatile and can be applied to a large number of scientific projects. If you want to learn more about it, find below a subset of our publications on this technology. Also, do not hesitate to contact us directly for any request for information, material and/or collaboration.
Selected publications
In order to develop our projects in an integrative biology approach, our team works collaboratively with research teams with complementary expertise to ours, in biology but also in chemistry. In addition, our technology and our know-how in the reconstitution of protein complexes by multi-expression leads us to develop collaborative projects in addition to our own projects. Some of our collaborations have been established for more than two decades, while other collaborations are more occasional. Our collaborations involve teams from the IGBMC, teams from Strasbourg, as well as teams at the national and international levels, as demonstrated by our publications.











Nature ; Volume: 505 ; Page: 648-653
Journal of Biological Chemistry ; Volume: 289 ; Page: 8989 - 8999
eLife ; Volume: 3 ; Page: 308-312
Journal of Molecular Biology ; Volume: 426 ; Page: 3442-3453
PLoS Pathogens ; Volume: 9
J Virol ; Volume: 87 ; Page: 8465-80
Current Opinion in Structural Biology ; Volume: 23 ; Page: 326-334
Cell ; Volume: 152 ; Page: 132--143
Journal of Biological Chemistry ; Volume: 287 ; Page: 27580-92
Protein Expression and Purification ; Volume: 80 ; Page: 8-16