
Stochastic Systems Biology of Gene Regulation
Stochastic Systems Biology of Gene Regulation

Biology is undergoing an exciting transformation. With the advent of single-cell genomics and large-scale imaging, we now have the tools to characterize every cell state in an organism. This opens the door to a systematic understanding of how cells work together in both health and disease. However, this progress presents exciting challenges for computational biology: new methods are needed to extract meaningful insights and generate reliable predictions from these massive datasets.
Deep learning has shown tremendous potential in addressing these challenges. Yet, the traditional "black-box" nature of deep learning limits its interpretability and, in turn, its predictive power. My team at the IGBMC is pushing the boundaries of computational biology by integrating biophysical modeling with generative deep learning to develop mechanistic and interpretable large-scale models of gene regulation. By incorporating prior biological knowledge of molecular interactions, dynamical systems, and statistical mechanics, we can better capture the gene expression dynamics that drive cellular behavior. Our models, trained on large-scale single-cell genomic data, pinpoint key regulators of specific gene programs and uncover complex, non-linear gene response functions that define combinatorial regulation.
Additionally, we have established a wet lab for conducting experiments in eukaryotic cell line models, enabling us to produce, analyze, and integrate single-cell sequencing and imaging data. This interdisciplinary approach allows us to generate data and create models that are not only powerful but also interpretable, leading to testable predictions based on causal mechanisms.
To explore the team's publications, please visit Google Scholar

Members
Researchers
Post-doctoral fellows
PhD students
Engineers
Current projects
- FateCycle. The cell cycle and cell differentiation are tightly interconnected processes that play a pivotal role in development and tissue regeneration. Studies have shown that specific phases of the cell cycle, such as G1, are critical windows for initiating cell-type-specific gene expression, linking cell cycle regulation to the activation of differentiation programs. In this project, we aim to characterize the interplay between cell cycle progression and cell differentiation in human induced pluripotent stem cells (hiPSCs) during definitive endoderm differentiation. We combine cutting-edge single-cell multiome sequencing techniques with advanced deep learning models to analyze gene expression and chromatin accessibility dynamics at unprecedented temporal resolution. The project seeks to disentangle cell cycle- and differentiation-dependent transcriptional and epigenetic mechanisms, estimate RNA biogenesis rates, and identify key transcription factors (TFs) driving these processes. By providing fundamental insights into the molecular dynamics of pluripotent cells, this research has the potential to improve differentiation protocols and advance regenerative medicine applications.
- CancerCycle. We aim to uncover the spatiotemporal regulation of the cell cycle within tumor microenvironments (TMEs) using spatial transcriptomics and a novel computational tool, CyclEM. By analyzing cell cycle dynamics and transitions to quiescence in various tumors, we investigate how TME features like cell density, composition, and morphology influence these processes. We integrate spatial and single-cell data to map gene expression and uncover transcription factor activities, using generative deep learning to model regulatory dynamics. Ultimately, we seek to reveal mechanisms underlying cancer progression, therapeutic resistance, and relapse, providing tools for personalized cancer therapies.
- ActiVAE. We aim to develop an interpretable variational autoencoder (VAE) to model gene regulation from single-cell transcriptomic data. While VAEs are powerful tools for clustering and denoising genomic data, their black-box nature limits the interpretability of latent variables. This project addresses this challenge by incorporating prior knowledge of gene interactions into the network structure of the VAE decoder, enabling latent variables to represent the activities of regulatory proteins. By identifying key regulators responsible for single-cell transcriptomes and applying optimal transport theory, the project seeks to predict the minimal regulatory changes required to transition between cell types. These advancements aim to provide insights into gene regulation and accelerate the development of new cell therapies.
- HiddenFoot. We have developed a novel approach that combines Single Molecule Footprinting (SMF) with a biophysical model based on Boltzmann-Gibbs statistics to study gene regulation at the single-DNA-molecule level. Traditional techniques like ChIP-seq and ATAC-seq provide only averaged views of nucleosome positioning and transcription factor (TF) binding, limiting insights into TF cooperativity and nucleosome dynamics. HiddenFoot overcomes these limitations by simultaneously determining nucleosome positioning, TF binding profiles, and RNA polymerase occupancy, significantly enhancing the interpretability of SMF data. This approach allows us to disentangle direct TF cooperativity from nucleosome-mediated effects and infer local TF concentrations genome-wide. Integrating these findings with chromatin 3D structure and live-cell imaging of transcriptional bursting dynamics will uncover new insights into regulatory hubs, transcriptional dynamics, and cooperativity, advancing our understanding of gene regulation in complex biological systems.
Resources
- DeepCycle is autoencoder to infer underlying cycling structure in scRNA-seq data and uncover gene regulation during the cell cycle in single cells.
github.com/MolinaLab-IGBMC/DeepCycle - FateCompass is a flexible computational framework to estimate dynamic stochastic trajectories of gene expression and transcription factor activities during cell-fate decision using single-cell RNA-seq data.
github.com/MolinaLab-IGBMC/fatecompass - GP-Tool is an user-friendly interphase to analyze the diffusion dynamic of particles in the living cells using Gaussian processes and fractional Brownian motion.
github.com/MolinaLab-IGBMC/GP-Tool. - FurierCycle is a novel computational approach to uncover fundamental rates of RNA metabolism and chromatin accessibility dynamics during the cell cycle. Results can be explored online.
github.com/MolinaLab-IGBMC/FourierCycle | molina.igbmc.science/FourierCycle.html - HiddenFoot is an efficient thermodynamic model to reveal transcription factor and nucleosome binding patterns on single DNA molecules using dynamical programming and stochastic gradient decent
github.com/MolinaLab-IGBMC/HiddenFoot
Awards and recognitions
2021 | USIAS Fellowship. University of Strasbourg Institute for Advanced Study, France.
2021 | Theory@EMBL Fellowship. EMBL Heidelberg, Germany.
2016 | IDEX Chair for Attracting Research. University of Strasbourg, France.
2016 | INRT LabEx Chair of Excellence. IGBMC. France.
2013 | Chancellor’s Fellowship. University of Edinburgh, UK.
2011 | SIB Young Bioinformatician Award. Swiss Institute of Bioinformatics, Switzerland.
Publications
-
2021
-
Regulation of transcription reactivation dynamics exiting mitosis
- Sergio Sarnataro
- Andrea Riba
- Nacho Molina
PLoS Computational Biology ; Volume: 17
-
Molecular Co-occupancy Identifies Transcription Factor Binding Cooperativity In Vivo
- Can Sönmezer
- Rozemarijn Kleinendorst
- Dilek Imanci
- Guido Barzaghi
- Laura Villacorta
- Dirk Schübeler
- Vladimir Benes
- Nacho Molina
- Arnaud Regis Krebs
Molecular Cell ; Volume: 81 ; Page: 255-267.e6
-
-
2020
-
First Responders Shape a Prompt and Sharp NF-κB-Mediated Transcriptional Response to TNF-α
- Samuel Zambrano
- Alessia Loffreda
- Elena Carelli
- Giacomo Stefanelli
- Federica Colombo
- Edouard Bertrand
- Carlo Tacchetti
- Alessandra Agresti
- Marco Bianchi
- Nacho Molina
- Davide Mazza
iScience ; Volume: 23 ; Page: 101529
-
Temporal specificity and heterogeneity of Drosophila immune cells
- Pierre Cattenoz
- Rosy Sakr
- Alexia Pavlidaki
- Claude Delaporte
- Andrea Riba
- Nacho Molina
- Nivedita Hariharan
- Tina Mukherjee
- Angela Giangrande
EMBO Journal ; Volume: 39
-
-
2019
-
Rfx6 promotes the differentiation of peptide-secreting enteroendocrine cells while repressing genetic programs controlling serotonin production
- Julie Piccand
- Constance Vagne
- Florence Blot
- Aline Meunier
- Anthony Beucher
- Perrine Strasser
- Mari Lund
- Sabitri Ghimire
- Laure Nivlet
- Céline Lapp
- Natalia Petersen
- Maja Engelstoft
- Christelle Thibault-Carpentier
- Céline Keime
- Sara Jimenez Correa
- Valérie Schreiber
- Nacho Molina
- Thue Schwartz
- Adèle de Arcangelis
- Gérard Gradwohl
Molecular metabolism ; Volume: 29 ; Page: 24-39
-
Visualization of Endogenous Transcription Factors in Single Cells Using an Antibody Electroporation-Based Imaging Approach
- Sascha Conic
- Dominique Desplancq
- Alexia Ferrand
- Nacho Molina
- Etienne Weiss
- Làszlò Tora
Imaging Gene Expression: Methods and Protocols, ; Page: 209-221
-
-
2018
-
Multiple inputs ensure yeast cell size homeostasis during cell cycle progression, Multiple inputs ensure yeast cell size homeostasis during cell cycle progression.
- Cecilia Garmendia-Torres
- Olivier Tassy
- Audrey Matifas
- Nacho Molina
- Gilles Charvin
eLife ; Volume: 7
-
Circadian clock-dependent and -independent posttranscriptional regulation underlies temporal mRNA accumulation in mouse liver
- Jingkui Wang
- Laura Symul
- Jake Yeung
- Cédric Gobet
- Jonathan Sobel
- Sarah Lück
- Pål Westermark
- Nacho Molina
- Felix Naef
Proceedings of the National Academy of Sciences of the United States of America ; Volume: 115 ; Page: E1916-E1925
-
Imaging of native transcription factors and histone phosphorylation at high resolution in live cells
- Sascha Conic
- Dominique Desplancq
- Alexia Ferrand
- Véronique Fischer
- Vincent Heyer
- Bernardo Reina San Martin
- Julien Pontabry
- Mustapha Oulad-Abdelghani
- Kishore Babu N.
- Graham Wright
- Nacho Molina
- Etienne Weiss
- Laszlo Tora
Journal of Cell Biology ; Volume: 217 ; Page: 1537-1552
-
Page 2 of 2
News
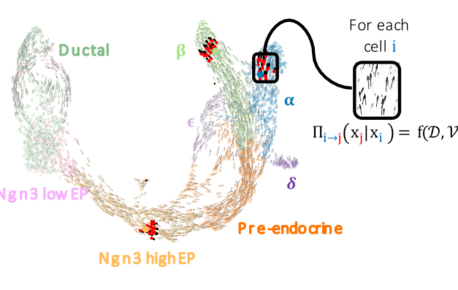
FateCompass: to identify and predict the decisive criteria of cell fate
Cell differentiation is a process regulated by gene expression through the action of transcription factors. Different transcription factors and…
Read more